
Abstract
SUMMARY: High-grade astrocytoma with piloid features (HGAP) is a recently identified brain tumor characterized by a distinct DNA methylation profile. Predominantly located in the posterior fossa of adults, HGAP is notably prevalent in individuals with neurofibromatosis type 1. We present an image-centric review of HGAP and explore the association between HGAP and neurofibromatosis type 1. Data were collected from 8 HGAP patients treated at two tertiary care institutions between January 2020 and October 2023. Demographic details, clinical records, management, and tumor molecular profiles were analyzed. Tumor characteristics, including location and imaging features on MR imaging, were reviewed. Clinical or imaging features suggestive of neurofibromatosis 1 or the presence of NF1 gene alteration were documented. The mean age at presentation was 45.5 years (male/female = 5:3). Tumors were midline, localized in the posterior fossa (n = 4), diencephalic/thalamic (n = 2), and spinal cord (n = 2). HGAP lesions were T1 hypointense, T2-hyperintense, mostly without diffusion restriction, predominantly peripheral irregular enhancement with central necrosis (n = 3) followed by mixed heterogeneous enhancement (n = 2). Two NF1 mutation carriers showed signs of neurofibromatosis type 1 before HGAP diagnosis, with one diagnosed during HGAP evaluation, strengthening the HGAP-NF1 link, particularly in patients with posterior fossa masses. All tumors were IDH1 wild-type, often with ATRX, CDKN2A/B, and NF1 gene alteration. Six patients underwent surgical resection followed by adjuvant chemoradiation. Six patients were alive, and two died during the last follow-up. Histone H3 mutations were not detected in our cohort, such as the common H3K27M typically seen in diffuse midline gliomas, linked to aggressive clinical behavior and poor prognosis. HGAP lesions may involve the brain or spine and tend to be midline or paramedian in location. Underlying neurofibromatosis type 1 diagnosis or imaging findings are important diagnostic cues.
ABBREVIATIONS:
- ATRX
- Alpha thalassemia/mental retardation syndrome X-linked
- DCE
- dynamic contrast-enhanced
- DMG
- diffuse midline glioma
- GTR
- gross-total resection
- HGAP
- high-grade astrocytoma with piloid features
- MAPK
- mitogen-activated protein kinase
- MC AAP
- methylation-class anaplastic astrocytoma with piloid features
- NF1
- neurofibromatosis type 1
- PA
- pilocytic astrocytoma
- PFS
- progression-free survival
- TPF3
- tumor protein p53
CNS tumor classification has incorporated several molecular markers and genetic mutations of prognostic value, as seen with the latest 5th edition of World Health Organization CNS tumor classification.1,2 For characterizing tumors with unusual morphologic features and overlapping characteristics on conventional histology, DNA methylation profiling is helpful for accurate classification and can alter the diagnosis in up to 12% of patients.3 Reinhardt et al4 used DNA methylation profiling on existing tumor data to identify a new subtype, initially termed “methylation-class anaplastic astrocytoma with piloid features (MC AAP),” later incorporated into the 5th edition as “High-Grade Astrocytoma with Piloid Features (HGAP).”5
HGAP is a rare tumor, primarily found in the posterior fossa (74%), but it can occur in supratentorial and spinal locations.4 It can emerge de novo with neurofibromatosis type 1 (NF1) and has also been reported to progress from a prior lower-grade glioma, often pilocytic astrocytoma (PA). Cimino et al6 identified 3 distinct epigenetic groups (g) of HGAPs based on DNA methylation: gNF1 (n = 18), g1 (n = 72), and g2 (n = 54), with median ages of 43.5, 47, and 32 years. Notably, gNF1 was strongly associated with a clinical diagnosis of NF1 (33.3%, P < .001), posterior fossa localization, neurofibromin 1 (NF1) hypermethylation, and reduced progression-free survival (PFS) (P < .058). The tumor histology varies and often displays frequent mitotic activity, elongated glial tumor cell processes, known as “piloid” features, and Rosenthal fibers or eosinophilic granular bodies. These tumors feature a unique epigenetic profile: mitogen-activated protein kinase (MAPK) pathway activation (often NF1, FGFR1, or BRAF alterations), cyclin-dependent kinase inhibitor 2 (CDKN2A/B) deletions (>70%), adenosine triphosphate–dependent helicase (ATRX) loss (50%), chromosome 19q loss (>50%), and occasional BRAF duplications. Some patients with HGAP may also exhibit a methylated MGMT promoter. HGAP may resemble glioblastoma and diffuse midline glioma (DMG) on imaging, posing a diagnostic challenge. The prognosis is intermediate between IDH-mutant gliomas and IDH wild-type glioblastomas with a 5-year survival rate of around 50%.4 Considering the rarity and uniqueness of HGAP, there is a lack of specific imaging biomarkers, which makes DNA methylation profiling the sole diagnostic method. Limited radiologic data are available, primarily derived from pathologic studies aimed at re-assigning CNS tumors to HGAP on the basis of DNA methylation.4,7,8 Herein, we present a dual-institution review of imaging findings in HGAP, supplementing molecular testing for precise diagnosis.
MATERIALS AND METHODS
The study was approved by the local institutional review boards at both institutions (Mayo Clinic Rochester, Minnesota Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania) with waived patient consent due to its retrospective nature. Institutional pathology records were searched between January 2020 and October 2023 for patients with an integrated HGAP diagnosis via histologic and genomic features (molecular DNA methylation classification and next-generation sequencing). We excluded patients in whom pathologists had determined that the DNA methylation confidence scores were below the agreed threshold (<0.9) for an HGAP diagnosis (as per the Bethesda classifier/Heidelberg classifier tool), implying a final reported non-HGAP integrated diagnosis.3 A total of 8 unique cases of HGAP were identified, 6 from the first and 2 from the second institution. We gathered clinical, demographic, imaging data and histopathologic, immunohistochemical, surgical, and postoperative details. All patients underwent a 3T whole-body MR imaging (Magnetom Skyra or Magnetom Prisma and Tim Trio; Siemens). The anatomic imaging protocol included axial 3D T1-weighted MPRAGE, axial T2 FLAIR, DWI, 3D SWI, and sagittal T1-sampling perfection with application-optimized contrasts by using different flip angle evolution (SPACE sequence; Siemens) imaging using standard parameters. The postcontrast T1-weighted images were acquired with the same parameters as the precontrast acquisition after administration of three-quarters of the standard dose (0.1 mmol/kg) of gadoterate meglumine (Dotarem; Guerbet) IV contrast agent, administered twice for dynamic contrast-enhanced (DCE) and DSC scans (a total of 1.5 full standard dose) using a power injector (Medrad, Idianola, PA). The DCE scans were performed after the first dose and included a fast 3D spoiled gradient-echo sequence (TR/TE = 5.09 ms/1.57 ms; flip angle = 23°; section thickness = 3.5 mm; FOV = 22 × 22 cm2; matrix size = 256 × 256; 28 slices per measurement, with 30 sequential measurements). The DSC scans included a T2*-weighted gradient-echo EPI sequence and were performed following the second dosage of contrast injection (TR/TE = 2000/45 ms; FOV = 22 × 22 cm2; matrix size = 128 × 128; in-plane resolution = 1.72 × 1.72 mm2; section thickness = 3 mm; bandwidth = 1346 Hz/pixel; flip angle = 90°; EPI factor = 128; echo spacing = 0.83; acquisition time = 3 minutes 10 seconds). Forty-five sequential measurements were taken per section. The injection rate for both scans was 5 mL/s, followed by a 20-mL saline flush at the same rate.
Four patients (P2, P6, P7, P8) had dynamic DSC, and 2 (P7, P8) had additional DCE-perfusion MR imaging. One patient with a spinal HGAP (P3) had whole-body FDG-PET/CT. Two neuroradiologists (N.S., G.B.) reviewed neuroimaging studies in consensus for location and MR imaging characteristics, including cysts, hemorrhage, and enhancement patterns (heterogeneous, necrotic with rim or patchy enhancement) alongside comprehensive pathologic and immunohistochemical analyses. Additionally, treatment and outcome/survival data were also collected. PFS was defined as the duration from the initial surgery to radiologic/clinical recurrence. Overall survival, the patient survival duration from the initial diagnosis, was available for 2 patients.
RESULTS
Patient Characteristics
The mean and median age at diagnosis was 45.5 (SD, 19.39 years; range, 19–71 years; male/female = 5:3) and 43 years, respectively. Common presenting symptoms included headache, weakness, and backache, with isolated cases of vocal cord paralysis (P5) and hearing loss (P6). Three patients (P3, P5, P6) showed clinical stigmata of NF1 (café-au lait spots and cutaneous/plexiform neurofibromas), with a positive family history of NF1 in 1 (P5) (Table).
| Patient No. | Sex | Age at Presentation (yr) | Location | Clinical Symptoms | Clinical/Imaging NF1 Features |
|---|---|---|---|---|---|
| 1 | M | 44 | Intramedullary T9-T10 | Midback pain | None |
| 2 | M | 42 | Midline pontomesencephalic and thalamic tumor, intraventricular tumor seeding; drop mets on follow-up MR imaging spine (C5-C6) | Confusion, fatigue, and nausea, lower backache | None |
| 3 | M | 74 | Intramedullary C5–T1 | Generalized weakness, difficulty walking, lower limb tingling and numbness | Scattered cutaneous/subcutaneous neurofibromas; multiple neurofibromas along cervicodorsal spine MR imaging; postsurgical resection of sciatic nerve plexiform NF1 |
| 4 | F | 38 | Left cerebellum | Gradually progressive headache and dizziness | None |
| 5 | M | 44 | Pontomedullary | Nausea, dyspnea, vocal cord paralysis | Suboccipital neurofibroma |
| 6 | F | 32 | Right pontine lesion extending into the right cerebellopontine angle, internal auditory canal, prepontine cistern | Bilateral hearing loss | Hyperpigmented macules and papules on chest; multiple plexiform neurofibromas |
| 7 | M | 19 | Midline diencephalic/thalamic region | Severe headache | None |
| 8 | F | 71 | Midline diencephalic/thalamic region | Syncope, loss of consciousness, urinary incontinence | None |
Note:—M indicates male; F, female; drop mets, leptomeningeal mets in spine.
Demographic and clinical features of the patient population with HGAP
Imaging Characteristics
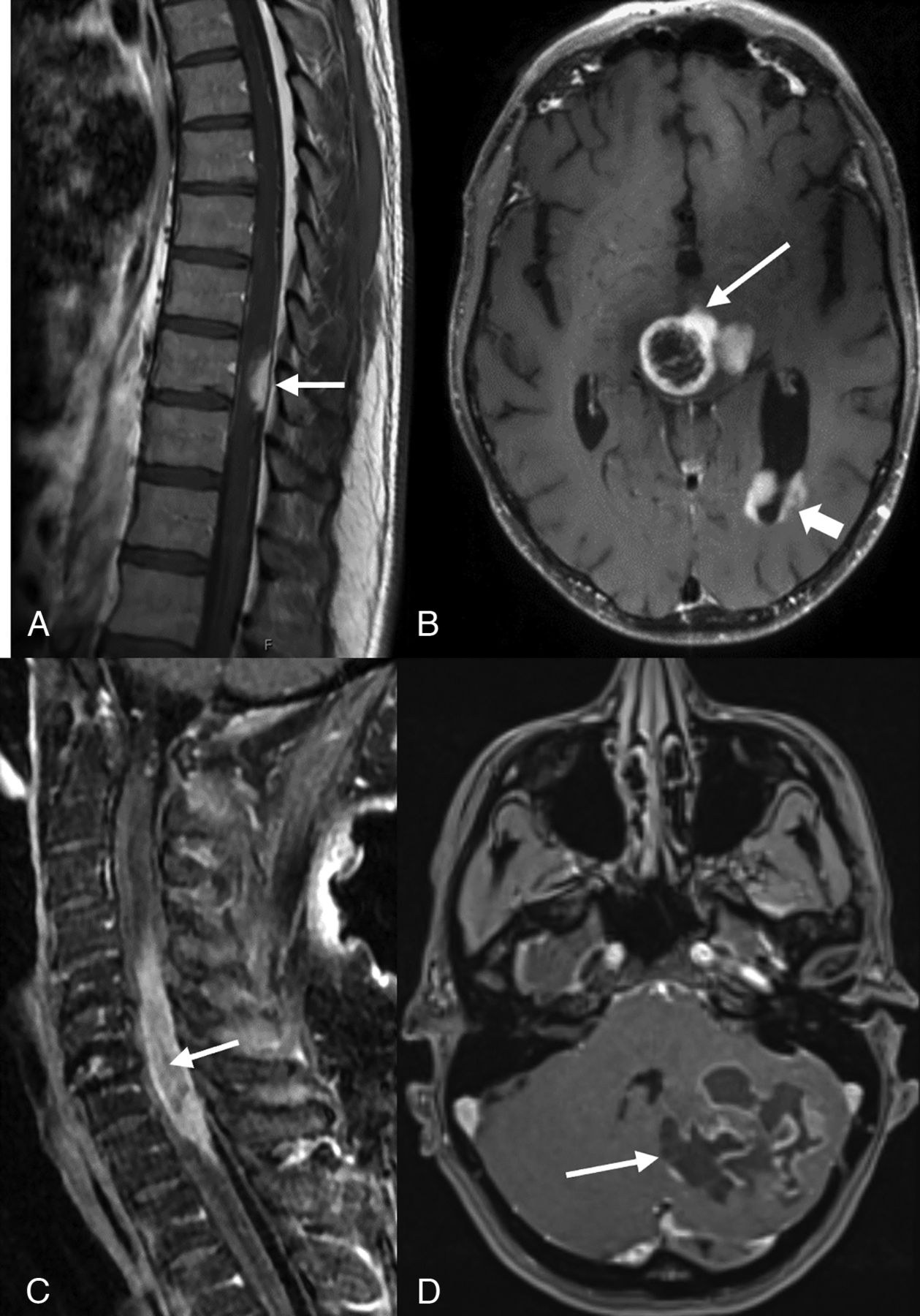
One-half of the lesions (4/8) were located in the posterior fossa (pontomedullary, cerebellar hemisphere, pons-cerebellopontine angle, and pontomesencephalic) followed by supratentorial (2/8) (midline diencephalic/thalamic region) and intramedullary (2/8) localization. Most lesions abutted the pial (4/8) or ependymal (3/8) surface. Spinal lesions in P1 (T9–T10) and P3 (C5–T1) were characterized by long-segment (>3 vertebral segments) intramedullary expansile tumors with a dorsal predilection. Spinal HGAPs exhibited exophytic components near the pial surface and were found to adhere to the pia during surgery (Fig 1A, -C). One of the posterior fossa pontine lesions (P6) had a predominant exophytic cerebellopontine component extending into the internal auditory canal and prepontine cistern (Fig 2B). HGAP lesions were predominantly T1 hypointense, T2/FLAIR/STIR hyperintense, and mostly without diffusion restriction, except that 1 lesion (P5) showed patchy diffusion restriction. Three HGAPs (P4, P7, P8) had intralesional hemorrhage (Online Supplemental Data). Most lesions exhibited enhancement, though the enhancement varied from minimal patchy ill-defined (P5, Online Supplemental Data) to plaquelike (P1, Fig 1A) to heterogeneous enhancement involving the tumor to a variable extent, without a dominant single pattern. Three diencephalic/thalamic lesions (P2, P7, P8) displayed peripheral, irregular enhancement with central necrosis (Figs 1B and 2C, -D). One intracerebellar HGAP (P4) exhibited a multicystic lesion with peripheral enhancement and areas of hemorrhage (Fig 1D and Online Supplemental Data).

Postcontrast-T1-weighted sagittal (A) image of a 44-year-old man with midback pain shows a well-defined enhancing lesion involving the dorsal aspect of thoracic cord at the T9–T10 level (arrow). Postcontrast T1-weighted axial image in a 42-year-old man (B) shows tumor with peripheral irregular enhancement at the level of thalamus (arrow) with infratentorial pontomesencephalic and cerebellar involvement (not shown) along with diffuse intraventricular tumor seeding (arrowhead). Postcontrast T1-weighted sagittal (C) image of a 74-year-old-man with weakness and difficulty walking shows an intramedullary heterogeneously enhancing tumor involving nearly the entire cord C5–T1 (arrow) along with cord expansion. Postcontrast T1-weighted-axial (D) image in a 38-year-old man demonstrates a large multicystic lesion with peripheral enhancement in the left cerebellum (arrow).

Postcontrast FLAIR axial MR image in a 44-year-old man (A) with dyspnea and vocal cord paralysis shows an expansile hyperintense lesion in the pontomedullary region with patchy ill-defined enhancement (not shown). Postcontrast T1-weighted axial MR images (not images). B, A 32-year-old woman presenting with hearing loss demonstrates a large, right pontine tumor extending into the right cerebellopontine angle, internal auditory canal, and prepontine cistern. Postcontrast T1-weighted axial MR images (C and D) demonstrate a midline, diencephalic/thalamic region tumor with rim enhancement in a 19-year-old man (C) and a 71-year-old woman (D).
MR perfusion in 4 patients (P2, P6, P7, P8) showed elevated perfusion parameters on DSC (relative CBV) and DCE (Ktrans, volume transfer coefficient; Ve, extravascular extracellular volume fraction; Vp, blood plasma volume fraction; Online Supplemental Data). One (P2) pontomesencephalic HGAP showed intraventricular seeding, progressing on follow-up MRIs with more extensive ependymal involvement (Fig 1B). This patient also exhibited dorsal intramedullary T2-hyperintense signal at the C5–C6 level with focal leptomeningeal enhancement developing 5 months after the initial MR imaging, consistent with leptomeningeal spread, a finding rarely reported with HGAP (Online Supplemental Data).9 One patient with a spinal HGAP (P3) showed increased tracer uptake on FDG-PET/CT, indicating the high-grade nature of the lesion (Online Supplemental Data). Figures 1 and 2, and the Online Supplemental Data depict HGAP imaging specifics.
Operative Details
Gross-total resection (GTR) was performed in 3 cases (P1, P3, P4); and subtotal resection, in 3 cases (P6, P7, P8) due to tumor vascularity. These patients received adjuvant chemoradiation (temozolomide, proton beam radiation therapy, and intensity beam radiation therapy) except one (P7), who died 1 month after surgery. Two patients did not undergo surgery due to multifocality (P2) and pontomedullary location (P5) and had only adjuvant chemoradiation (Online Supplemental Data).
Histopathologic and Molecular Characteristics
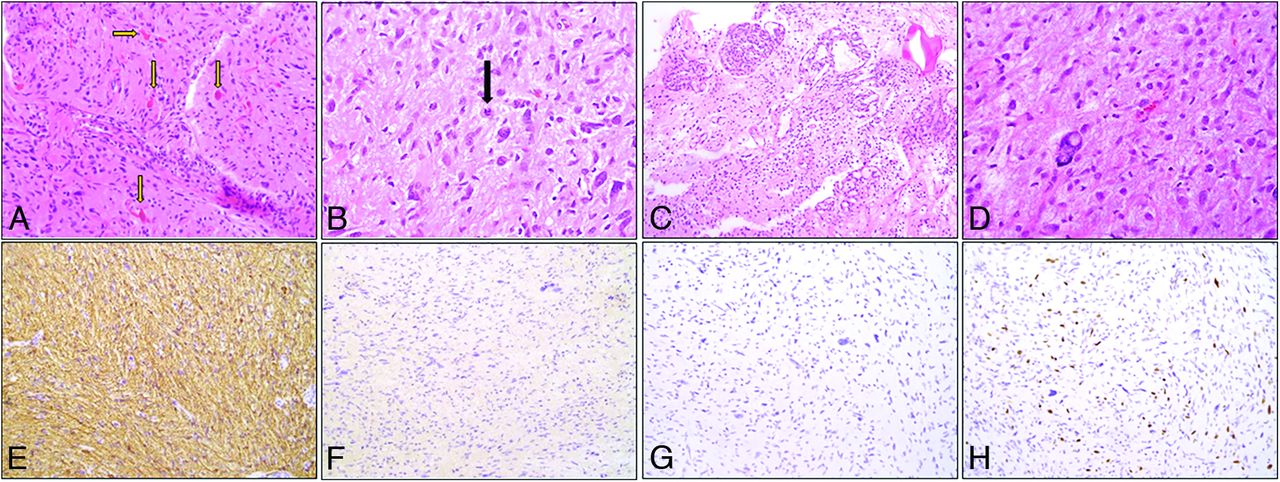
Histopathology showed a moderately cellular glial tumor with variable mitotic activity and nuclear atypia. Some tumors showed glomeruloid vasculature. Rosenthal fibers or eosinophilic granular bodies were seen in a minority. Microhemorrhage was present in 3 HGAPs (P6, P7, P8). Cystic changes were present in 3 HGAPs (P4, P7, P8). Next-generation sequencing data were obtained from various institutions with different gene region coverage. The most common genetic alterations were CDKN2A/B homozygous deletion (7/8, 87.5%), ATRX mutation (87.5%), MGMT promoter methlation (6/8, 75%), and NF1 gene alteration (5/8) (62%). Six HGAP cases had co-occurring CDKN2A/B homozygous deletion and ATRX mutations. Other observed mutations included TP53 (3/6, 50%), RB1 loss (2/3), BRCA2 loss (1/3), and SETD2 (1/3). All tumors were IDH1/2 wild-type. Histone H3 mutations, such as the common H3K27M typically seen in DMGs, linked to aggressive clinical behavior and poor prognosis, were not detected in our cohort.10 A representative histopathologic and immunohistochemical profile is shown in Fig 3 and the Online Supplemental Data. NF1 mutations were found in 62.5% (P1, P2, P3, P5, P6), of which 2 (P3, P6) had prior clinical (cutaneous neurofibromas) and imaging (plexiform neurofibroma) NF1 stigmata. These patients later developed intraspinal and posterior fossa HGAP after 6 months and 6 years of initial NF1 diagnosis, respectively. One patient (P5) was diagnosed with cutaneous neurofibromas during HGAP work-up along with an NF1 family history. The overall pathologic NF1 mutation rate is 92.1% (70/76) in individuals with NF1 identified by next-generation sequencing.11 CSF analysis for malignant cells was negative in all cases. Intraspinal HGAP (P3) was a highly cellular glioma with nuclear anaplasia, infiltrative growth, and high mitotic activity, with additional p53 overexpression that lacked microvascular proliferation or tumor necrosis.

Morphologic and immunohistochemical profile of HGAP. A, H&E, 200× stained section shows a moderately cellular tumor with abundant Rosenthal fibers and eosinophilic globular bodies (arrows). B, H&E (400×) stained section highlights cells with hairlike processes imparting a piloid appearance (arrow denotes a mitotic figure). C, H&E-stained section (100×) demonstrates glomeruloid vasculature often associated with HGAP. D, H&E-stained section (400×) shows bizarre atypia seen in scattered cells. E, Immunohistochemical (200×) stains. Glial fibrillary acidic protein shows diffuse positivity. F, IDH1-R132H with absence of staining, G, ATRX stain demonstrates loss. H, Ki-67.
Follow-Up
Mean and median follow-ups were at 17.4 and 23 months, respectively. While the treatments varied, the PFS was available in P1 (3 months) and P4 (26 months). One patient (P3) had a residual tumor after surgery without response. One patient (P1) underwent a re-resection after 15 months of initial GTR for recurrence. Six patients were alive at the time of the last follow-up, and 2 (P2, P7) died with 1–12 months of overall survival (Online Supplemental Data).
DISCUSSION
HGAP is a new entity within circumscribed astrocytic gliomas, often resembling glioblastoma rather than PA with survival comparable with that of grade IV IDH-mutant astrocytoma.5 DNA methylation profiling is performed to exclude HGAP, especially in atypical cerebellar gliomas (with ATRX loss or CDKN2A/B homozygous deletion) and HGAP-resembling tumors (like high-grade gliomas in NF1).6 Reinhardt et al8 found that molecular analysis reclassified approximately one-third of initially diagnosed glioblastoma cases as HGAP.12 In a study by Lucas et al,13 advanced molecular diagnosis reclassified 14 NF1-associated gliomas as HGAP, further supporting the NF1 and HGAP link. Although the exact HGAP incidence in NF1 remains uncertain, a confirmed NF1 diagnosis should alert clinicians to the potential HGAP risk.
The NF1 gene is a tumor-suppressor gene mutated in individuals with NF1. NF1 encodes the neurofibromin 1 protein, which negatively regulates the Ras/MAPK pathways via Ras inhibition. NF1 mutations are prevalent in treatment-resistant gliomas and serve as supportive diagnostic biomarkers for HGAP and rosette-forming glioneuronal tumors.14,15 Patients with NF1 are at a higher risk of developing low-grade gliomas. NF1 also predisposes to high-grade gliomas, with their prevalence being 10–50 times higher than in the general population. Romo et al16 reported a nearly 300-fold higher prevalence of non-optic pathway gliomas in patients with NF1 (3.2%) compared with the general population (0.01%), with a median survival of 24 months. Nonoptic gliomas in adults with NF1 often have an aggressive clinical course, further underscoring the need to understand the pathobiology of NF1-associated gliomas better.16 NF1-related gliomas resembling HGAP had worse outcomes than the NF1-associated PA but fared better than sporadic IDH wild-type glioblastoma.13 A substantial GB-HGAP overlap necessitates early HGAP detection because these HGAP cases often have a better prognosis and access to targeted therapies.13 NF1-mutated tumors may respond to mammalian target of rapamycin (mTOR) and MAPK inhibitors, such as FDA-approved everolimus, temsirolimus, and emerging complex inhibitors.15 This possibility raises the question of whether brain/spine MR imaging screening is advisable for those with NF1, but the lack of large-scale studies on HGAP prevalence in NF1 prevents a conclusive answer.
One-half of HGAP lesions in our cohort involved the posterior fossa, followed by equal (25%) spinal cord and midline diencephalic/thalamic region involvement. Reinhardt et al4 and Bender et al7 identified the posterior fossa as the primary location in 74% and 66%, respectively. Spinal cord involvement varied between 7% (5/83)4 and 33% (2/6).7 We found 5 HGAPs (P1, P3, P5, P6, P8) in the midline, aligning with the observations of Romo et al16 indicating increased midline involvement in NF1-associated tumors. Among these 5 cases, GTR was performed in 2 cases (P1, P3), with one (P1) achieving a PFS of 3 months, matching the findings of Romo et al of limited GTR success in NF1-associated tumors. While tumor-imaging descriptions for HGAP in the literature are sparse, they generally exhibit variable T1-weighted hypointensity, T2-weighted hyperintensity, and heterogeneous enhancement. Diffusion restriction was generally absent. In our cohort, HGAP lesions closely imitated glioblastoma with variable heterogeneous enhancement and increased perfusion metrics. Intramedullary HGAPs were expansile with a dorsal predilection, and one (P3) demonstrated FDG-avidity. This imaging profile is similar to the findings of Bender et al7 in their 6-case series, including radiotracer avidity on O-(2-[18F] fluoroethyl)-l-tyrosine PET/CT in 2 cases. Focal areas of signal intensity are common in patients with NF1 and may sometimes be challenging to differentiate from tumors. Perfusion-weighted MR imaging offers the potential to detect tumor-related neoangiogenesis, aiding in the differentiation to determine whether focal areas of signal intensity represent a low-grade tumor.17 One of our patients with HGAP (P2) demonstrated ependymal and intraspinal leptomeningeal spread, indicating aggressiveness and a poor prognosis. Leptomeningeal or dural spread of HGAP is rarely reported, except for a recent study that identified leptomeningeal spread in a confirmed case of HGAP with a 7-year interval to metastasis and at the time of initial diagnosis in a possible HGAP. Adult high-grade astrocytic tumor types, including HGAP, are capable of leptomeningeal or dural spread.9
MGMT promoter methylation status is crucial for therapy and prognosis. Unmethylated status predicts a poor response to alkylating chemotherapy and worse prognosis.5,18 In our cohort, all except 1 patient received alkylating chemotherapy. We detected MGMT promoter methylation in 62.5% (5/8) of patients, higher than that reported by Bender et al7 (33%, 2/6) and Reinhardt et al4 (45%, 38/83). Four of our 5 cases of NF1 gene mutation also had co-occurring ATRX and CDKN2A/B mutations. Patients with IDH wild-type astrocytic gliomas with CDKN2A/B deletions and ATRX mutations generally have a better clinical outcome than patients with IDH wild-type glioblastoma, but the significance in NF1 is unclear.4 Both CDKN2A/B and ATRX mutations were common as reported by Lucas et al,13 who also noted MAP kinase pathway mutations (NF1, FGFR-1, BRAF genes). ATRX mutations were found in 87.5% (7/8) of our cases, slightly higher than previously reported in 45%–60% of cases.4,7 Additionally, CDKN2A/B mutation was identified in 87.5% of our cases (7/8), previously reported in 80%–100% of cases.4,7 ATRX, a chromatin remodeling protein, preserves genomic stability and often co-occurs with IDH mutations, potentially improving survival in low-grade gliomas, especially when lacking 1p/19q codeletions.19 While ATRX alterations are characteristic of IDH-mutant astrocytoma, they are also frequently found in the IDH wild-type HGAP in conjunction with homozygous deletion of CDKN2A/B. This loss of CDKN2A/B generally indicates a higher grade and poorer prognosis, especially compared with PA.4,5 HGAP is diagnosed solely by methylation profiling; however, the loss of ATRX on immunohistochemistry is helpful, especially with concurrent homozygous loss of CDKN2A/B to suggest this entity. In cases without ATRX mutation, TERT can be mutated (these are mutually exclusive events).6
In 2 cases (P7 and P8) FGFR1 fusion was detected, which transforms primary astrocytes into highly proliferating midline gliomas.20,21 TP53 mutations were identified in 50% (3/6) of the patients in our study, a rate higher than the 5.4% reported in cases of HGAP by Cimino et al.6 TP53, a tumor-suppressor gene, is involved in DNA damage response, cell cycle arrest, and apoptosis, with its mutations enhancing cell survival and chemoresistance.5,22 BRAF V600E mutations were evaluated in 50% of cases, and all findings were negative. BRAF fusions are typically linked to World Health Organization grade 1 PA pediatric posterior fossa cases, showing a favorable outcome compared with supratentorial PAs in adults.4 One patient with HGAP (P4) exhibited a rare genetic profile of NTRK2 fusion, loss of P16 expression, a SETD2 mutation, lack of CDKN2A/B deletion, and MGMT promoter methylation. Considering these characteristics, this case was contemplated for potential inclusion in a future trial involving NTRK-targeted therapy (entrectinib) in the event of disease progression. The significance of these mutations remains uncertain; however, they merit further investigation in future studies. SETD2 mutations have been reported in high-grade hemispheric gliomas in older children23 and adults with cerebellar glioblastomas.24 Loss-of-function mutations in SETD2 result in a deficiency of trimethylated histone H3K36, which appears specific to high-grade tumors. The nonsense mutation in SETD2 in addition to a KIAA1549-BRAF fusion, contributes to the aggressive clinical course of this tumor.23,24
HGAP can be challenging to distinguish from PA, DMGs, and glioblastoma on imaging. PAs, typically found in children, are low-grade circumscribed gliomas with good prognosis (10-year survival rate >95%). These are strongly associated with NF1 and BRAF-KIAA1549 fusion/duplication. Most sporadic PAs arise from the cerebellum while affecting the optic pathway in patients with NF1. However, in adults, PAs, mostly supratentorial, exhibit a more aggressive clinical course, with a 50% 5-year survival rate and rare BRAF V600E alterations.25 Radiologically, they show variable appearances, ranging from large cystic lesions with enhancing nodules to solid lesions with intense enhancement (∼95%), calcification (20%), and hemorrhage.26 The transformation of PA into a high-grade glioma is uncommon, especially in cases involving BRAF fusion, while about 20% of HGAPs with BRAF fusion suggest a possible link to earlier PAs or shared biologic traits.8,20 Diffuse midline glioma (H3K27-altered) often occurs in pediatric patients without significant sex variation. It is characterized as an expansile, diffusely infiltrative lesion primarily in the thalamus, brainstem, and spinal cord, presenting as slightly T1 hypointense and T2 hyperintense on MR imaging with lesser enhancement, higher diffusion restriction, and perfusion. H3K27-altered DMGs have a poor prognosis, despite their histopathologic grade, mandating H3K27M detection and genetic analysis for definitive identification.27,28 HGAP, prevalent in adults, associated with NF1, often displays high T2 signal and variable peripheral enhancement, and lacks diffusion restriction.7 Both tumors share a dismal prognosis and are managed with surgery, radiation, and chemotherapy.7,29 Both HGAP and glioblastoma, IDH wild-type high-grade CNS tumors, share similar imaging features and poor prognosis, requiring DNA methylation profiling for accurate differentiation. Glioblastoma, the most frequent primary brain tumor, typically manifests as a heterogeneous mass in the cerebral hemisphere, displaying irregular peripheral enhancement, central necrosis, diffusion restriction, extensive peritumoral edema, and glioblastoma histologic criteria (microvascular proliferation and necrosis).7
Our study did not reveal significant imaging differences between HGAPs with and without NF1 mutations, except that spinal HGAPs were linked to NF1. Patient outcomes may be influenced by a complex interplay of factors, including NF1 mutations, genetic modifiers, extent of tumor involvement, resectability, and treatment response. Neither our current study nor existing literature indicate any notable prognostic differences between HGAPs with and without NF1 mutations; however, individual cases may differ due to unique genetic and molecular traits. Our study is constrained by a small patient sample, retrospective design, and varying next-generation sequencing data availability across institutions with differing gene region coverage. Nevertheless, this imaging-focused review adds valuable insight into the clinical, genetic diversity, and imaging characteristics of HGAP, particularly in NF1-associated HGAPs.4,8 Targeted therapy may be pivotal due to frequent MAP kinase pathway gene alterations.6
CONCLUSIONS
Radiologists should consider HGAP as a reasonable differential diagnosis in patients with high-grade/aggressive-appearing glial neoplasms in patients with NF1, especially with midline or paramedian localization. Nevertheless, the small sample size of our study due to HGAP rarity underscores the need for future larger studies or meta-analyses to assess the strength of the NF1-HGAP association.
Footnotes
Disclosure forms provided by the authors are available with the full text and PDF of this article at www.ajnr.org.
References
- Received October 31, 2023.
- Accepted after revision December 21, 2023.
- © 2024 by American Journal of Neuroradiology




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.