
Abstract
Summary: A 9-year-old Haitian girl presented initially with monocular blindness and an isolated temporal arteritis, confirmed by angiographic studies and temporal artery biopsy findings. CT and MR studies of the intracranial circulation showed only an enlarged, dense superficial temporal artery. Systemic workup revealed a mildly elevated erythrocyte sedimentation rate, mild changes in white and red blood cells, and a remote history of sensorineural hearing loss. Pathologic examination of the biopsy specimen narrowed the differential diagnosis to giant cell temporal arteritis and polyarteritis nodosa. Treatment with corticosteroids alone failed, and the child returned 1 month later with severe systemic illness and encephalopathy. MR studies showed multiple cortical and subcortical foci of increased T2 signal, and gyriform enhancement on T1-weighted images. Renal and mesenteric arteriograms showed innumerable tiny aneurysms at branch points in small and medium-sized vessels, typical of polyarteritis nodosa. We found no previous reports of this initial presentation in the pediatric population for either polyarteritis nodosa or giant cell temporal arteritis.
Polyarteritis nodosa (PAN) is a disease of small or medium-sized muscular arteries with typical involvement of the abdominal viscera, heart, CNS, and skin. Other sites, such as the superficial temporal artery, are involved rarely (1–3). PAN predominates in young adults, but can occur in all age groups. When PAN presents in an atypical location and atypical age group, the diagnosis becomes less straightforward. The exclusion of other vasculitides with overlapping clinical, radiologic, and pathologic manifestations may become difficult.
We report a case of a 9-year-old Haitian girl with PAN who presented with temporal arteritis and monocular blindness. We believe this case is unique, as we were unable to find any previous reports of this presentation in a child. Furthermore, this constellation is more commonly observed in giant cell temporal arteritis (GCTA) (1). The youngest reported patient with biopsy-proved GCTA with this presentation, however, was a 35-year-old man (4). Although a benign form of temporal arteritis, called juvenile temporal arteritis (JTA), has been reported in children, this rare disease differs both clinically and pathologically from the case we present (5, 6).
We review the clinical, radiologic, and pathologic features of the three vasculitides (PAN, JTA, and GCTA) considered in the differential diagnosis and emphasize the importance of integrating these features so as to arrive at the correct diagnosis in cases of atypical disease presentations.
Case Report
A 9-year-old Haitian girl visiting in the United States had a history of sensorineural hearing deficit since age 4. She was admitted to the emergency department with 1- to 2-month history of intermittent fever (100–102°F), chills, blindness in the left eye, intermittent left temporal and periorbital headaches, acute left temporal scalp tenderness, and left-sided periorbital tenderness. Additionally, she had a more recent onset (several days) of periumbilical aching abdominal pain, low back pain, aching distal lower extremity pain, odynophagia, and anorexia. Also noted was a 3-month history of intermittent confusion, memory deficits, and intermittent development of a whole-body rash during crying.
Physical examination revealed swelling of the left upper eyelid with orbital fullness; a tender, enlarged, pulsatile left temporal artery; left pupillary afferent defect with paradoxical pupillary dilatation during contralateral light stimulation; left-sided photophobia; ophthalmoscopically noted optic nerve atrophy; an asymmetric smile; and bilateral lateral gaze nystagmus. Abnormal laboratory results included an elevated erythrocyte sedimentation rate (57 mm/h), RBC elevation (5.07 × 106 μL), WBC depression (4.3 K/μL), and mean corpuscular hemoglobin depression (25 pg/mL). Urine analysis, serum amylase, and liver enzyme levels were normal. Screening chemistries were otherwise noncontributory. Serology tests (purified protein derivative, rapid plasma reagin, HIV antibody, hepatitis A antibody test, hepatitis B surface-antigen and antibody, varicella-zoster antibody, stool ova and parasite examination, blood and urine cultures) for various infectious processes (tuberculosis, syphilis, HIV, hepatitis A, hepatitis B, varicella-zoster, enteric pathogens, and parasites) were negative. A bone marrow aspirate was unremarkable.
A noncontrast CT scan of the brain showed mild generalized atrophy, with asymmetry of the superficial temporal arteries, left larger than right (Fig 1A). The enlarged, dense, superficial temporal artery measured 54 HU, consistent with blood clot, but not calcification. An MR study showed only diffuse, mild cortical atrophy, with no focal findings. A left common carotid arteriogram showed multiple foci of short- and long-segment narrowing in the proximal left superficial temporal artery, with a prominent beaded appearance more distally (Fig 1B). A right common carotid injection showed significantly less involvement of the right superficial temporal artery, consisting of mild focal segment stenosis (not shown). No definite intracranial vascular abnormalities were seen. An arch aortogram was normal.

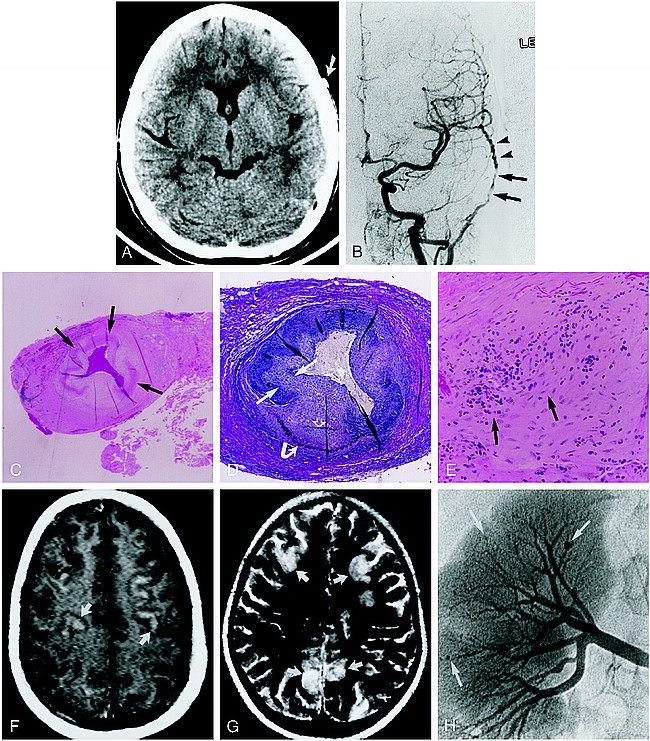
9-year-old girl with left-sided monocular blindness and frontal headaches associated with tenderness over the temporal region.
A, Noncontrast CT scan shows an enlarged and dense left temporal artery (arrow). Bone window algorithm (not shown) and HU measurement showed no evidence of calcification.
B, Digital subtraction angiogram, anteroposterior view from a left common carotid injection, shows focal, long-segment stenosis (arrows) and beading (arrowheads) of the left superficial temporal artery.
C–E, Biopsy findings of the left temporal artery. Low-power view (C) shows focal irregularity and near circumferential fibrinoid necrosis in the biopsy specimen (arrows). Note the clot within the lumen of the vessel (hematoxylin-eosin; original magnification ×10). Cross section of the superficial temporal artery (D) shows marked intimal hyperplasia (straight arrows) with disruption of the internal (open arrow) and external (curved arrow) elastic membranes (elastin; original magnification ×40). High-power view of the vessel media (mid wall) (E) shows a mixed cellular lymphocytic-predominant infiltrate (arrows) (original magnification ×100).
F and G, Contrast-enhanced T1-weighted (800/20/4) (F) and T2-weighted (2000/200/1) (G) axial brain MR images (obtained after clinical relapse, including bilateral blindness, mental status changes, and abdominal pain) show multiple peripheral focal areas of contrast enhancement and T2 prolongation involving the frontoparietal cortex (arrows).
H, Digital renal angiogram obtained 1 day after the MR study shows multiple peripheral microaneurysms (arrows), consistent with systemic PAN.
Results of a left temporal artery biopsy showed intimal hyperplasia, disruption of the internal and external elastic membranes, a mixed cellular (lymphocytic predominate) infiltrate, and fibrinoid necrosis, consistent with either PAN or GCTA (Fig 1C–E). After correlating biopsy results, clinical findings, and radiologic findings, a tentative diagnosis of GCTA was made by the consulting rheumatologist, with differential considerations given to PAN and JTA. The latter was excluded clinically (see Discussion).
The patient was started on intravenous steroids, and rapidly improved, with several weeks of remission, before relapsing. She was readmitted 4 weeks later with acute binocular blindness, microscopic hematuria, mental status changes, and elevated CSF protein. A contrast-enhanced MR examination showed acute, multifocal, bilateral, cortically based foci of gyriform enhancement and increased T2 signal (Fig 1F and G). Abdominal angiography showed multiple microaneurysms in the kidneys (Fig 1H) and mesenteric arterial system, predominantly at small-vessel bifurcations in a pattern characteristic of PAN. A revised final diagnosis of PAN was made by the consulting rheumatologist.
Discussion
Histologically, medium-sized arteries are composed of three well-defined layers: the tunica intima, the tunica media, and the tunica adventitia. The intima is composed of connective tissue, which in turn is composed of collagen, proteoglycans, elastin, and matrix glycoproteins. Endothelial cells line the blood vessel lumen, defining the inner border of this layer. The intima is separated from the media by the internal elastic lamina, which is fenestrated. The media consists predominantly of concentric circular or spiral smooth muscle layers with sparse fine elastic fibers, and is delineated at its outer boundary by the external elastic membrane. The adventitia consists of connective tissue, elastic and nerve fibers, and nutrient vessels (vasa vasorum).
All three disorders, PAN, GCTA and JTA, can affect the medium-sized vessels, such as the superficial temporal artery, although their histopathology differs. Classic PAN can affect both medium or small arteries in any organ other than the lung. Our case demonstrates many of the typical histologic findings of PAN (Fig 1C–E). Three stages of vessel changes are often seen (7). Acute lesions are characterized by fibrinoid necrosis (Fig 1C), which can involve part or all of the vessel wall. A mixed cellular infiltrate (Fig 1E) is often present in and around the vessel wall. Affected segments can develop aneurysmal dilatation and the lumen can thrombose. In addition to necrosis, subacute lesions show evidence of healing, such as fibroblastic proliferation (Fig 1E) and infiltration by macrophages and by plasma cells. The adventitia is sometimes involved, producing gross vascular nodularity. Chronic lesions result in markedly thickened, fibrotic walls. Elastic tissue stains usually show fragmentation of the internal elastic lamina (Fig 1D). In typical biopsy specimens, lesions in multiple stages of evolution are commonly present.
The angiographic features of PAN include segmental stenosis (resulting pathologically from intimal hyperplasia, cellular infiltration, and fibrinoid necrosis) and beading (resulting from tightly spaced, irregular, short, narrowed segments with intervening normal or aneurysmal vessels) (Fig 1B).
However, histologic, angiographic, and cross-sectional imaging findings overlap between PAN and the other vasculitides (GCTA and JTA) considered in this case. Histologic features of GCTA usually fall into three patterns: granuloma replete with giant cells and with a fragmented internal elastic lamina, a nonspecific white cell infiltrate throughout the arterial wall, or intimal fibrosis without disruption of the internal elastic membrane. Giant cells are present in only half to two thirds of cases (7, 8). Features shared between GCTA and PAN include intimal thickening, inflammation, and disruption of the elastic lamina. Although fibrinoid necrosis has been described as a rare feature in GCTA (7), it is not widely recognized as such, and its presence should strongly weight the diagnosis in favor of PAN.
JTA is an unusual inflammatory condition of external carotid artery branches, first described by Lie et al in 1975 (5). It is different from classic temporal arteritis both clinically and histopathologically (5, 6). It can present unilaterally or bilaterally, as either a painless or a tender nodule in the temporal region (5, 6). Patients with JTA are often asymptomatic, without a history of preceding or concurrent systemic illness. This disease usually occurs unilaterally as either a painless or a tender nodule in the temporal region, and has a benign course as compared with GCTA. The high-dose corticosteroid management required for GCTA is unnecessary. Histopathologic features of JTA include a non-giant cell granulomatous inflammation of the arteries with intimal proliferation and microaneurysmal disruption of the media (5, 6). To our knowledge, fibrinoid necrosis has not been described in this entity.
While the initial angiographic findings in our case correlate well with the histopathology of PAN, they are also considered classic for GCTA. In fact, when isolated to the superficial temporal artery, this appearance was once thought sensitive and specific for GCTA (9). As in our case, however, this appearance has been reported in other diseases (10). To our knowledge, angiographic criteria have not been described for JTA.
The CT and MR findings in our patient also overlap with those findings previously reported in GCTA. Figure 1A shows an enlarged, dense superficial temporal artery with a density of 54 HU; it was not well seen on images filmed with a bone window algorithm. This appearance is consistent with blood clot, which was identified histologically (Fig 1C). Similar findings have been demonstrated previously in GCTA (11), although the dense temporal artery in the previous study contained calcifications (based on HU criteria and histologic findings). While typical for findings occurring in PAN (12), the intracranial lesions seen on MR images obtained during our patient's second admission (Fig 1F and G) have also been rarely mimicked by GCTA (13, 14). We were unable to determine the pathogenesis of the focal areas of gyriform enhancement and of increased T2 signal. Although our patient showed significant neurologic changes at the time of admission, no clinical evidence of seizure activity was apparent. Diffusion-weighted images were not obtained, and the patient returned to Haiti shortly after the study was performed, precluding follow-up. In one previous series, similar changes were thought to represent focal areas of infarction (12).
Given that the histopathologic and angiographic findings of vasculitis may overlap in a pediatric patient, it is necessary to proceed with caution in arriving at a final diagnosis. Even after correlation with clinical findings, the composite findings may not completely clarify the diagnosis. In 1990, the American College of Rheumatology outlined a classification system for several of the major vasculitides (15). As most rheumatologic diseases lack pathognomonic features, this classification system was initiated to establish uniformity in differentiating various types of vasculitis. The classification system is, however, somewhat limited, as the full spectrum of disease manifestations is not included. As a result, the authors state that their classification system is not equivalent to diagnostic criteria, and may not be appropriate for diagnosis of an individual patient (15). The present case is an example of this point.
The criteria for PAN as put forth by the American College of Rheumatology include any three of the following: weight loss greater than or equal to 4 kg, livedo reticularis, testicular pain or tenderness, myalgias, mononeuropathy or polyneuropathy, diastolic blood pressure greater that 90 mm Hg, elevated blood urea nitrogen or serum creatinine levels, presence of hepatitis B reactants in serum, arteriographic abnormality, and presence of granulocyte or mixed leukocyte infiltrate in an arterial wall biopsy (16). The initial presentation of our patient met more (four) than the minimal three criteria required for classification as PAN.
The criteria established by the American College of Rheumatology for classification of the traditional form of GCTA include new onset of localized headache, temporal artery tenderness or decreased temporal artery pulse, elevated erythrocyte sedimentation rate, arterial biopsy findings of necrotizing arteritis characterized by a predominance of mononuclear cell infiltrates or a granulomatous process with multinucleated giant cells, and age greater than or equal to 50 years at disease onset (8). Other than the age criteria, the case we present satisfies more than the minimum three criteria needed for classification as GCTA.
Arriving at a final diagnosis in our case required integration of clinical, radiologic, and pathologic findings. The pattern of vascular involvement allowed differentiation between the two diseases. GCTA most frequently involves the medium-sized extracranial arteries, and only rarely involves the aorta and its primary branches, the kidneys, the lungs, abdominal viscera, and skin (8, 17, 18). PAN, on the other hand, commonly affects both medium-sized and small arteries in these organs. Distinction between these two diseases is clinically important, since GCTA can often be treated with high-dose steroids alone, and has a more benign course. PAN is a more severe disease that requires both steroids and cytotoxic therapy.
Conclusion
Temporal arteritis is a rare disorder among the pediatric population. When the initial presentation involves temporal arteritis, the clinical aggressiveness of the disease helps to distinguish JTA (relatively benign) from more aggressive entities. In our case, monocular blindness precluded the diagnosis of JTA. When temporal arteritis is aggressive, laboratory and clinical data help to distinguish infectious from noninfectious pathologic mechanisms. When no evidence exists of an infectious pathogenesis, we suggest that careful attention be paid to radiologic/pathologic correlation, and that PAN be considered the most likely diagnosis in the presence of histologic evidence of fibrinoid necrosis of the vessel wall.
Footnotes
↵1 Address reprint requests to Robert J. Bert, MD, PhD, Department of Radiology, Boston Medical Center, 88 E Newton St, Boston, MA 02118.
References
- Received October 14, 1997.
- Accepted after revision May 8, 1998.
- Copyright © American Society of Neuroradiology



{kind=link}