
Abstract
BACKGROUND AND PURPOSE: Among cognitively healthy older individuals, the relationship among the 2 hallmark proteins of AD (Aβ and τ APOE ε4) and neurodegeneration is not well-understood. Here, we investigated the relationship between Aβ, p-τ, and APOE ε4 on longitudinal brain atrophy in preclinical AD.
MATERIALS AND METHODS: We examined 107 cognitively healthy older adults who underwent longitudinal MR imaging and baseline lumbar puncture. Within the same linear mixed-effects model, we concurrently investigated main and interactive effects between the APOE ε4 genotype and CSF Aβ1–42, CSF p-τ and CSF Aβ1–42, and the APOE ε4 genotype and CSF p-τ on entorhinal cortex atrophy rate. We also examined the relationship of APOE ε4, CSF p-τ, and CSF Aβ1–42 on the atrophy rate of other AD-vulnerable neuroanatomic regions.
RESULTS: The full model with main and interactive effects demonstrated a significant interaction only between CSF p-τ and CSF Aβ1–42 on entorhinal cortex atrophy rate, indicating elevated atrophy with time in individuals with increased CSF p-τ and decreased CSF Aβ1–42. The APOE ε4 genotype was significantly and specifically associated with CSF Aβ1–42. However, the interaction between the APOE ε4 genotype and either CSF Aβ1–42 or CSF p-τ on entorhinal cortex atrophy rate was not significant. We found similar results in other AD-vulnerable regions.
CONCLUSIONS: On the basis of our findings and building on prior experimental evidence, we propose a model of the pathogenic cascade underlying preclinical AD in which APOE ε4 primarily influences the pathology of Alzheimer disease via Aβ-related mechanisms, and in turn, Aβ-associated neurodegeneration occurs only in the presence of p-τ.
ABBREVIATIONS:
- Aβ
- amyloid-β
- AD
- Alzheimer disease
- APOE ε4
- ε4 allele of apolipoprotein E
- HC
- healthy controls
- p-τ
- phospho-τ181p
- SE
- standard error of the mean
Converging biochemical, molecular, and genetic evidence indicates that Aβ plays a central role in the neurodegenerative process underlying AD.1 The presence of Aβ initiates loss of dendritic spines and synapses2 and contributes to the dysfunction of neuronal networks.3 Reports based on mouse models suggest that multiple factors influence Aβ-associated toxicity. The ε4 allele of APOE ε4, the most important genetic risk factor for late-onset AD,4 accelerates the onset of Aβ deposition into plaques5 and decreases the transport of Aβ across the blood-brain barrier.6 Reductions in τ, another hallmark protein of AD pathology, protect against Aβ-induced neuronal dysfunction,7 while the presence of τ potentiates Aβ-associated synapotoxicity.8
In humans, evidence from genetic-at-risk cohorts and neuropathologic findings in clinically healthy older individuals suggest that the pathobiologic process underlying AD begins years before the onset of cognitive deficits or dementia symptoms.9 Biomarker studies in cognitively asymptomatic older adults have demonstrated significant relationships between structural MR imaging measures of brain atrophy and CSF Aβ levels,10⇓–12 enabling identification of clinically healthy individuals who may be in a presymptomatic or preclinical stage of AD.13
Recent evidence from our laboratory indicates that in clinically healthy older individuals and those with mild cognitive impairment, Aβ-associated volume loss occurs only in the presence of p-τ.14 However, it is unknown whether APOE ε4 and CSF p-τ concurrently modulate the effect of CSF Aβ on longitudinal brain atrophy in preclinical AD. In this study, we investigated whether concurrent interactions between decreased CSF Aβ1–42 and APOE ε4 and between decreased CSF Aβ1–42 and increased CSF p-τ are associated with increased brain atrophy in cognitively healthy older individuals.
Materials and Methods
Selection of participants and analysis methods for MR imaging and CSF biomarkers are briefly summarized here, with details provided in the On-line Appendix.
We evaluated participants who were clinically diagnosed at baseline as cognitively and clinically healthy controls (global Clinical Dementia Rating = 0) from the Alzheimer Disease Neuroimaging Initiative. A total of 115 cognitively healthy older individuals had undergone longitudinal MR imaging, CSF lumbar puncture, and APOE ε4 genotyping. Of these individuals, we restricted our analyses to those participants (n = 107) with quality-assured baseline and at least 1 follow-up MR imaging (6 months to 3.5 years; 10% with 6-month follow-up, 15% with 12-month follow-up, 34% with 23-month follow-up, and 41% with 36-month follow-up) available as of December 2011. We classified all participants on the basis of the presence (“carriers”) and absence (“noncarriers”) of at least 1 APOE ε4 allele (Tables 1 and 2). Using recently proposed CSF cutoffs,15 we also classified all participants on the basis of high (>23 pg/mL, “positive”) and low (<23 pg/mL, “negative”) p-τ levels, and on low (<192 pg/mL, “positive”) and high (>192 pg/mL, “negative”) Aβ1–42 levels (Tables 1 and 2).
Demographic, clinical, and imaging data for all older HC in this study, as assessed by P-τ and Aβ status
Demographic, clinical, and imaging data for all older HC in this study, as assessed by APOE ε4 and Aβ status
We examined 417 T1-weighted MR images. We performed quantitative surface-based analysis of all MR images by using an automated region-of-interest labeling technique16 and primarily focused on entorhinal cortex, a medial temporal lobe region that is selectively affected in the earliest stages of AD.17⇓⇓–20 To additionally investigate neuroanatomic regions that are involved in the later stages of the disease process17,18 and to minimize multiple comparisons, we averaged longitudinal volume change in the temporal pole, parahippocampal gyrus, inferior temporal gyrus, banks of the superior temporal sulcus, inferior parietal lobule, amygdala, and hippocampus to create an “AD-vulnerable” region of interest (Fig 1). Using an automated method developed in our laboratory,21 we assessed longitudinal subregional change in gray matter volume (atrophy) on serial MR images.

3D representations of the neuroanatomic regions examined in the current study (only 1 hemisphere is shown). All of the neocortical regions are visible in the lateral (A) and medial (B) views of the gray matter surface, and the 2 non-neocortical regions (ie, the hippocampus and amygdala, C) are visible in the coronal view of a T1-weighted MR image. Regions illustrated in red constitute the AD-vulnerable region of interest (for further details please see text).
We asked whether p-τ and APOE ε4 independently influence Aβ-associated neurodegeneration. To investigate this question, we examined the main and interactive effects of CSF Aβ1–42 and APOE ε4, and CSF Aβ1–42 and CSF p-τ on entorhinal cortex atrophy rate in a mixed-effects model, covarying for the effects of age and sex, specifically
 Here, Δv is entorhinal cortex atrophy (millimeters) and Δt is the change in time from baseline MR imaging (in years). Using the same linear mixed-effects framework, we also investigated the main and interactive effects of CSF Aβ1–42 and APOE ε4, and CSF Aβ1–42 and CSF p-τ on the atrophy rate in the AD-vulnerable region of interest.
Here, Δv is entorhinal cortex atrophy (millimeters) and Δt is the change in time from baseline MR imaging (in years). Using the same linear mixed-effects framework, we also investigated the main and interactive effects of CSF Aβ1–42 and APOE ε4, and CSF Aβ1–42 and CSF p-τ on the atrophy rate in the AD-vulnerable region of interest.
Results
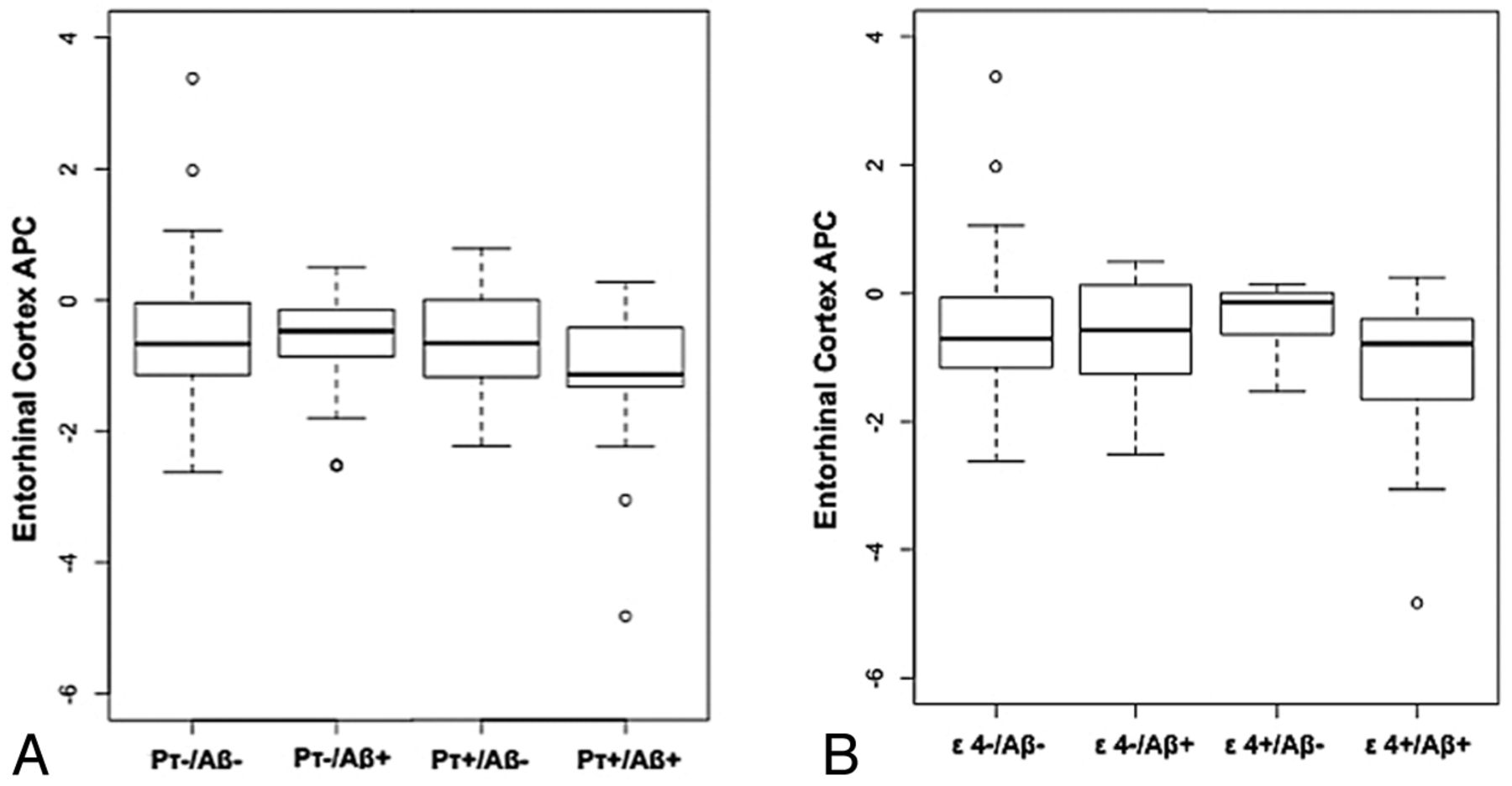
Results from the full model with both interactive terms showed that the interaction between CSF Aβ1–42 and CSF p-τ status on entorhinal cortex atrophy rate was significant (β5 = −0.39, SE = 0.14, P = .005), indicating elevated atrophy with time in individuals with positive CSF p-τ and positive CSF Aβ1–42 status (Fig 2A) as previously reported.14 In contrast, the interaction between CSF Aβ1–42 and APOE ε4 on entorhinal cortex atrophy rate was not significant (β4 = −0.17, SE = 0.18, P = .35). With both interaction terms in the model, the main effects of APOE ε4, CSF Aβ1–42 status, and CSF p-τ status were not significant. Follow-up analyses demonstrated that positive CSF Aβ1–42 status was associated with an elevated entorhinal cortex atrophy rate only among CSF p-τ–positive individuals (β-coefficient = −0.32, SE = 0.11, P = .008). There was no association between positive CSF Aβ1–42 status and entorhinal cortex atrophy rate among CSF p-τ–negative individuals (β-coefficient = 0.10, SE = 0.08, P = .23) (Fig 2A). There was no association between positive CSF Aβ1–42 status and entorhinal cortex atrophy rate either among APOE ε4 carriers (β-coefficient = −0.11, SE = 0.19, P = .58) or noncarriers (β-coefficient = −0.02, SE = 0.08, P = .76) (Fig 2B).

Box-and-whisker plots for all healthy control participants illustrating entorhinal cortex atrophy rate, measured as annualized percentage change (APC) based on CSF p-τ and CSF Aβ status (A) and ε4 genotype and CSF Aβ status (B). For each plot, thick black lines show the median value. Regions above and below the black line show the upper and lower quartiles, respectively. The dashed lines extend to the minimum and maximum values with outliers shown as open circles. As illustrated in A, the pτ+/Aβ+ HC demonstrate the largest cortical atrophy rate (ie, more negative percentage change). In comparison as noted in B, the ε4+/Aβ+ HC show equivalent rates of atrophy compared with the other groups.
Similar results were obtained when examining the association of CSF protein and APOE ε4 status on the atrophy rate in the AD-vulnerable region of interest: The interaction of CSF Aβ1–42 and CSF p-τ status on the AD-vulnerable region-of-interest atrophy rate was significant (β-coefficient = −0.34, SE = 0.11, P = .002), but the interaction of CSF Aβ1–42 and APOE ε4 was not (β-coefficient = −0.15, SE = 0.14, P = .28). None of the main effects of APOE ε4, CSF Aβ1–42 status, and CSF p-τ were significant with both interaction terms in the model. Follow-up analyses demonstrated that positive CSF Aβ1–42 status was associated with an elevated AD-vulnerable region-of-interest atrophy rate among CSF p-τ–positive individuals (β-coefficient = −0.30, SE = 0.09, P = .001) but not among CSF p-τ–negative individuals (β-coefficient = 0.03, SE = 0.07, P = .61). There was no association between positive CSF Aβ1–42 status and atrophy rate in the AD-vulnerable region of interest either in APOE ε4 carriers (β-coefficient = −0.19, SE = 0.13, P = .09) or noncarriers (β-coefficient = −0.06, SE = 0.07, P = .38).
We also examined the possibility that APOE ε4 modulates AD-associated neurodegeneration via p-τ–related mechanisms. Using the same linear mixed-effects model framework described above, we concurrently examined the main and interactive effects of APOE ε4 and CSF p-τ, CSF Aβ1–42 and APOE ε4, and CSF Aβ1–42 and CSF p-τ on the atrophy rate of entorhinal cortex and the AD-vulnerable region of interest. We did not find a significant interaction between APOE ε4 and CSF p-τ either on the atrophy rate of entorhinal cortex (β-coefficient = −0.04, SE = 0.18, P = .78) or the AD-vulnerable region of interest (β-coefficient = 0.19, SE = 0.15, P = .18). Most important, even within this triple interaction model, the only significant effect was the interaction between CSF Aβ1–42 and CSF p-τ on the atrophy rate of entorhinal cortex (β-coefficient = −0.38, SE = 0.15, P = .01) and the AD-vulnerable region of interest (β-coefficient = −0.41, SE = 0.12, P = .001).
Finally, although our results did not demonstrate a significant interaction between APOE ε4 and CSF Aβ1–42 on longitudinal brain atrophy among HC, we examined whether the presence of APOE ε4 is associated with decreased CSF Aβ1–42 and increased CSF p-τ by using a generalized linear model, covarying for age and sex, specifically
 We found a significant relationship between APOE ε4 status and positive CSF Aβ1–42 status (β-coefficient = 0.40, SE = 0.07, P = 4.82 × 10−7), indicating increased Aβ deposition in ε4 carriers. In contrast, there was no relationship between APOE ε4 carriers and positive CSF p-τ status (β-coefficient = 0.05, SE = 0.09, P = .55).
We found a significant relationship between APOE ε4 status and positive CSF Aβ1–42 status (β-coefficient = 0.40, SE = 0.07, P = 4.82 × 10−7), indicating increased Aβ deposition in ε4 carriers. In contrast, there was no relationship between APOE ε4 carriers and positive CSF p-τ status (β-coefficient = 0.05, SE = 0.09, P = .55).
Discussion
In this study, we show that in cognitively healthy older individuals, though the presence of the ε4 allele is specifically associated with Aβ deposition, APOE ε4 does not affect Aβ-associated volume loss. In contrast, we found that p-τ modulates Aβ-associated neurodegeneration in clinically healthy individuals, as previously reported.14 These findings, in conjunction with recent experimental observations,22,23 support a conceptual model of the pathogenic cascade underlying preclinical AD (Fig 3), in which APOE ε4 primarily influences Alzheimer pathology via Aβ-related mechanisms; and in turn, Aβ-associated neurodegeneration occurs only in the presence of p-τ. This model provides a representation of the disease process that can be assessed with currently validated biomarkers, not a comprehensive framework of all pathologic processes occurring in the earliest stages of AD. As such, it can be expanded to include future findings such as mechanistic details regarding the effect of genetic susceptibility loci on AD-associated neurodegeneration.

A conceptual model of AD-associated neurodegeneration in the preclinical phase of the disease process based on data from our mixed-effects models (please see text for details). The thickness of the arrows illustrates the magnitude of effect. The circle with a dot inside illustrates an interactive effect, the plus sign illustrates a positive effect, and X illustrates no significant effect.
These findings provide important insights into the preclinical stage of AD. Although several studies in cognitively asymptomatic older individuals have demonstrated a significant relationship among APOE ε4 genotype, Aβ deposition, and neurodegeneration,10⇓–12,24⇓–26 there has been limited evaluation of the role of p-τ in modulating these relationships. Our findings indicate that in clinically healthy older individuals, Aβ deposition by itself, either in ε4 carriers or noncarriers, is not associated with volume loss; the presence of p-τ represents a critical link among the APOE ε4 genotype, Aβ deposition, and neurodegeneration. Consistent with prior reports,27,28 our results illustrate that the ε4 allele primarily affects AD in an indirect fashion via Aβ. In contrast, these findings do not support a role for APOE ε4 either in affecting intracranial p-τ levels or modulating AD pathology via p-τ–related mechanisms.
From a quantitative neuroimaging perspective, our results demonstrate the feasibility of using automated MR imaging–based measures of longitudinal brain atrophy as an in vivo biomarker even at the preclinical stage of the disease process. Building on prior neuroimaging studies in cognitively healthy older adults,10⇓–12,24⇓–26 these findings indicate that volume loss can be detected in older individuals testing positive for both Aβ and p-τ. Furthermore, the pattern of atrophy detected in this study is consistent with previous neuropathologic studies demonstrating neuronal loss within entorhinal cortex in the earliest stages of AD.19,20 Taken together, these findings suggest that the regionally specific volume loss occurring in a subset of cognitively healthy older adults is neuropathologically consistent with early AD.
This study has limitations. One concern is that CSF biomarkers provide an indirect assessment of amyloid and neurofibrillary pathology and may not fully reflect the pathologic processes underlying Alzheimer disease. Another limitation is that we primarily focused on the APOE ε4 genotype and CSF biomarkers of the 2 pathologic hallmarks of AD. Additional genetic and cellular markers may also interact with Aβ to predict neurodegeneration in cognitively healthy elders. Finally, the individuals examined here may represent a group of highly selected, generally healthy older adults who are motivated to participate in research studies. These findings therefore need to be further validated on an independent community-based cohort of older individuals who would be more representative of the general older population.
Clinically, these results indicate that a biomarker profile evaluating both Aβ and p-τ may better identify those older individuals who are at an elevated risk of progressing to eventual dementia than either biomarker by itself. Consistent with prior clinical observations from our laboratory,29 our current findings suggest that early intervention trials should take into account both the p-τ and Aβ status of participants because older individuals with increased CSF p-τ and decreased CSF Aβ1–42 levels are likely to have significantly elevated rates of volume loss compared with individuals with normal CSF p-τ and decreased CSF Aβ1–42 levels. Finally, in addition to the current emphasis on Aβ, our findings identify the need for developing novel therapies that target APOE- and τ-related processes. It is likely that a complex interplay between multiple genetic and molecular entities determines AD pathogenesis.30,31 As such, targeting “upstream” events such as neuronal lipids and cholesterol transporters that interact with APOE in ε4 carriers with normal AD biomarker levels as well as “downstream” events such as τ phosphorylation and aggregation in older individuals with both decreased CSF Aβ1–42 and increased CSF p-τ levels may represent additionally beneficial treatment strategies.
Conclusions
We show that in cognitively healthy older individuals, p-τ modulates the effect of Aβ on neurodegeneration. In contrast, although the presence of the ε4 allele is specifically associated with Aβ deposition, APOE ε4 does not influence Aβ-associated volume loss. These findings provide important insights into the pathogenic cascade underlying preclinical AD and illustrate the importance of examining both Aβ and p-τ in secondary prevention trials.
Acknowledgments
The authors thank Drs William Bradley and John Hesselink for helpful input on this article.
Footnotes
Disclosures: Rahul S. Desikan—RELATED: Other: American Society of Neuroradiology Cornelius Dyke Award. Linda K. McEvoy—RELATED: Grant: NIH,* Comments: My efforts in this manuscript were supported by grants from the National Institute on Aging, grant numbers R01AG031224, K01AG029218; spouse is CEO of Cortechs Labs, Inc. Dominic Holland—OTHER RELATIONSHIPS: patent pending for quantitative anatomic regional change methodology, filed through University of California at San Diego technology-transfer office; coinventors D.H. and D.M.A. James B. Brewer—UNRELATED: Board Membership: Lilly Biomarker Business Unit, Bristol-Myers-Squibb, Comments: Advising on the use of imaging in Alzheimer clinical trials and clinical practice, Grants/Grants Pending: General Electric Medical Foundation,* Janssen Alzheimer Immunotherapy, Stock/Stock Options: Cortechs Labs Inc. Paul S. Aisen—RELATED: NIH;* UNRELATED: Consultancy: Elan Corporation, Wyeth, Eisai Inc, Bristol-Myers Squibb, Eli Lilly and Company, NeuroPhage, Merck & Co, Roche, Amgen, Abbott, Pfizer Inc, Novartis, Bayer, Astellas, Dainippon, Biomarin, Solvay, Otsuka, Daiichi, AstraZeneca, Janssen, Medivation Inc, Theravance, Cardeus, Anavex, Grants/Grants Pending: research grants from NIH,* Foundation for the National Institutes of Health,* Alzheimer's Association,* Baxter,* Pfizer,* Lilly.* Ole A. Andreassen—RELATED: Grant: Research Council of Norway,* Comments: travel expenses for and support for sabbatical at University of California at San Diego, UNRELATED: Other: pharmaceutical companies, Comments: I have received honoraria for lectures from pharmaceutical companies involved in medication for psychiatric disorders (Lundbeck, Lilly, Janssen, GlaxoSmithKline, MSD). Some of these companies may have medication for Alzheimer disease, but I am not aware of it. Bradley T. Hyman—RELATED: NIH grant to the Alzheimer Disease Research Center.* Reisa A. Sperling—RELATED: Grant: Regents of the University of California, San Diego,* Comments: 4-ADNI-PIB/ADC-029 (“PET Imaging of Brain Amyloid”), UNRELATED: Consultancy: Bayer,* Bristol-Myers-Squibb,* Eisai,* Janssen,* Kyowa Hakko Kirin,* Pfizer,* Roche,* Satori,* Avid, Comments: Avid is unpaid, Grants/Grants Pending: NIH.* Anders Dale—RELATED: NIH/National Institute on Aging,* Comments: grant No. R01AG031224, UNRELATED: Grants/Grants Pending: NIH,* Patents (planned, pending, or issued): University of California at San Diego,* Massachusetts General Hospital/Harvard Medical School,* Comments: I am an inventor of multiple pending and issued patents on MRI acquisition and analysis methods, filed through University of California at San Diego or Massachusetts General Hospital/Harvard Medical School, Stock/Stock Options: Cortechs Labs Inc, Comments: I am a founder and equity holder of Cortechs Labs, Inc and also serve on its Scientific Advisory Board. The terms of this arrangement have been reviewed and approved by the University of California, San Diego, in accordance with its conflict-of-interest policies, Other: GE Healthcare,* Comments: I am also the Principal Investigator of a research agreement between GE Healthcare and University of California at San Diego. *Money paid to the institution.
This research was supported by grants from the National Institutes of Health (R01AG031224, K01AG029218, K02 NS067427, T32 EB005970). Data collection and sharing for this project were funded by the Alzheimer Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Abbott, Alzheimer's Association, Alzheimer's Drug Discovery Foundation, Amorfix Life Sciences Ltd, AstraZeneca, Bayer HealthCare, BioClinica Inc, Biogen Idec Inc, Bristol-Myers Squibb Company, Eisai Inc, Elan Pharmaceuticals Inc, Eli Lilly and Company, F. Hoffmann-La Roche Ltd and its affiliated company Genentech Inc, GE Healthcare, Innogenetics, N.V., IXICO Ltd, Janssen Alzheimer Immunotherapy Research & Development LLC, Johnson & Johnson Pharmaceutical Research & Development LLC, Medpace Inc, Merck & Co Inc, Meso Scale Diagnostics LLC, Novartis Pharmaceuticals Corporation, Pfizer Inc, Servier, Synarc Inc, and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by National Institutes of Health grants P30 AG010129 and K01 AG030514.
Data used in preparation of this article were obtained from the ADNI data base (http://adni.loni.ucla.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in the analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.ucla.edu/wpcontent/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Rahul S. Desikan received the 2012 Cornelius G. Dyke Memorial Award, given to a trainee or junior faculty member in neuroradiology for excellence as demonstrated in a paper, which represents original unpublished research in some aspect of neuroradiology.
Indicates open access to non-subscribers at www.ajnr.org
References
- Received May 12, 2012.
- Accepted after revision June 11, 2012.
- © 2013 by American Journal of Neuroradiology
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.