
Abstract
SUMMARY: In 2021, the World Health Organization released an updated classification of CNS tumors. This update reflects the growing understanding of the importance of genetic alterations related to tumor pathogenesis, prognosis, and potential targeted treatments and introduces 22 newly recognized tumor types. Herein, we review these 22 newly recognized entities and emphasize their imaging appearance with correlation to histologic and genetic features.
ABBREVIATIONS:
- AT/RT
- atypical teratoid/rhabdoid tumor
- cIMPACT-NOW
- Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy-Not Official WHO
- GFAP
- glial fibrillary acidic protein
- MAPK
- mitogen-activated protein kinase
- NEC
- not elsewhere classified
- NOS
- not otherwise specified
- PFA and PFB
- posterior fossa ependymoma groups A and B
- WHO
- World Health Organization
- WHO CNS5
- World Health Organization Classification of Tumors of the Central Nervous System, fifth edition
- IDH
- isocitrate dehydrogenase
The World Health Organization (WHO) published the World Health Organization Classification of Tumors of the Central Nervous System, fifth edition (WHO CNS5).1 WHO CNS5 builds on the fourth edition, published in 2016, and the recommendations of the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy-Not Official WHO (cIMPACT-NOW),2 which produced 7 updates between 2018 and 2020.3⇓⇓⇓⇓⇓-9
The continued discovery of pathologically relevant molecular markers, along with an improved understanding of secondary alterations in tumor biology and clinical course, has led to recognition of 22 new tumor types, in addition to nomenclature changes to the existing classification. Three provisional entities are included, which appear clinicopathologically distinct but await additional studies before full acceptance.1
Despite the increasing realization of the altered molecular profile and clinical course, the imaging data on the newly recognized entities remain scarce, mostly confined to case reports and small case series.10⇓-12 Herein, we present a consolidated review of the WHO CNS5 new tumor types with emphasis on the common imaging findings. A brief review of the general changes to the tumor taxonomy, nomenclature, and grading system is also presented.
Immunohistochemistry and Molecular Markers
Basic histology has been the backbone of previous WHO classifications of >100 known CNS tumors. Under these classifications, however, there was marked interobserver variability and poor differentiation of tumors with diverse biologic behavior. Immunohistochemistry provided major insights into the cellular markers of tumor phenotype and stronger correlations with tumor behavior, resulting in improved standardization. This information has been in routine use for more than a decade with continual improvements and discovery of new immunohistochemical stains. In 2016, for the first time, molecular markers were used in addition to histology for the classification of CNS tumors. WHO CNS5 makes a substantial addition of specific genetic markers to immunohistochemistry and histology. Epigenetic markers, particularly alterations in DNA methylation, have also been added. These have proved immensely valuable not only for diagnosis but also for prognosis and treatment guidance.
During the past decade, DNA methylation profiling has emerged as a powerful tool for research, which has started making its way into the classification system. At present, it can assess the methylation status of 850,000 cytosine-guanine sites across the human genome with huge data sets matched through standardized controls, providing great precision in tumor identification. Using these techniques, the German Cancer Research Center and Heidelberg University have provided a reference cohort for almost all known tumor entities (www.molecularneuropathology.org). Machine learning tools can easily and accurately match a sample cohort with the references in the data base.13,14 At present, 4 of the newly recognized tumors use unique methylation profiles as part of their defining characteristics (high-grade astrocytoma with piloid features, diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters, and posterior fossa ependymoma, groups A and B [PFA and PFB]).
Tumor Taxonomy and Nomenclature
WHO CNS5 recognizes the variability in the need for molecular marker–based diagnoses. Some tumors have molecular characteristics that enable a complete diagnosis, whereas other tumors do not require a molecular approach for diagnosis. Thus, the current hybrid taxonomy is thought to represent an intermediate stage preceding even more precise future classifications. The only pertinent changes to the current taxonomy are “type” replacing “entity” and “subtype” replacing “variant.”1
WHO CNS5 attempts to make nomenclature more consistent and simpler per the 2019 cIMPACT-NOW Utrecht meeting recommendations.9 WHO CNS5 uses simplified tumor names and only includes location, age, or genetic modifiers with established clinical utility. Nevertheless, the nomenclature changes are not uniformly applied because several historical terms are deeply ingrained in the literature (eg, medulloblastoma, myxopapillary ependymoma, pleomorphic xanthoastrocytoma), and name changes could be substantially disruptive to clinical care and scientific experiments. Gene and protein nomenclature has been updated to bring consistency across other existing guidelines.
CNS Tumor Grading
Two noteworthy changes to the grading system include the use of Arabic instead of Roman numerals and the use of tumor grades within types. For example, anaplastic astrocytoma, which was WHO grade III, is no longer a tumor type. Rather, an isocitrate dehydrogenase (IDH)-mutant tumor is now categorized as grade 2, 3, or 4 based on a combination of histologic and molecular information. Both changes ensure more nomenclature uniformity across classification systems of non-CNS tumors. Additionally, the use of tumor grades within types allows more flexibility, while at the same time emphasizing biologic similarity within tumor types.1
Not Otherwise Specified and Not Elsewhere Classified
Not otherwise specified (NOS) implies a lack of or failure to obtain available molecular, histologic, or genetic information, which limits making a specific diagnosis. Not elsewhere classified (NEC) refers to cases in which the diagnostic testing has been successful but the results do not readily conform to a standard diagnosis under WHO CNS5. Both modifiers are primarily meant to alert the oncologist to either a lack of complete work-up (NOS) or lack of a standard diagnosis despite adequate work-up (NEC).
Bone and Soft-Tissue Tumors
WHO CNS5 attempts to align the classification of mesenchymal nonmeningothelial tumors with the WHO classification of bone and soft-tissue tumors. Tumors that overlapped both classification systems but were rarely encountered in the CNS (eg, leiomyoma) were removed.1 Additionally, 3 newly recognized mesenchymal tumors were added to WHO CNS5: intracranial mesenchymal tumor, FET-CREB fusion-positive, CIC-rearranged sarcoma, and primary intracranial sarcoma, DICER1-mutant.1
The following sections describe the WHO CNS5 newly described tumor types and are summarized in the Online Supplemental Data. For a general review of WHO CNS5, the readers are referred to an excellent review by Osborn et al.15
Diffuse Astrocytoma, MYB- or MYBL1-Altered
This WHO grade 1 tumor is 1 of 4 low-grade pediatric tumors that require molecular differentiation from one another due to their similar, nonspecific, low-grade histologic characteristics.1 The defining feature of this tumor is structural variation, such as fusion, rearrangements, or amplification, involving MYB or MYBL1, which are transcriptional regulators for cellular proliferation and differentiation.16 IDH and H3 are wild-type by definition.6 The median age at diagnosis is 5 years (range, 0–26 years), and there is no sex predilection.17 Reflecting the histopathology, the imaging features are nonspecific but typical of a low-grade glioma with an infiltrative, heterogeneously T2-hyperintense, nonenhancing, non-diffusion-restricting mass. The cerebral cortex is the most common location, followed by supratentorial white matter/deep gray nuclei, then the brainstem (Fig 1).17

Diffuse astrocytoma, MYB- or MYBL1-altered. A cortical T2 FLAIR (A) and T2 (B) hyperintense mass is noted within the right precentral gyrus (arrows) with low T1 signal and no enhancement (C). Histopathology reveals mildly hypercellular white matter with vaguely hypocellular areas and atypical glial cells (D), which are positive for GFAP (E) and synaptophysin (F). Chromosomal microarray analysis revealed a gain of 8q13.1q21.3, disrupting MYBL1. This is a WHO grade 1 tumor.
Polymorphous Low-Grade Neuroepithelial Tumor of the Young
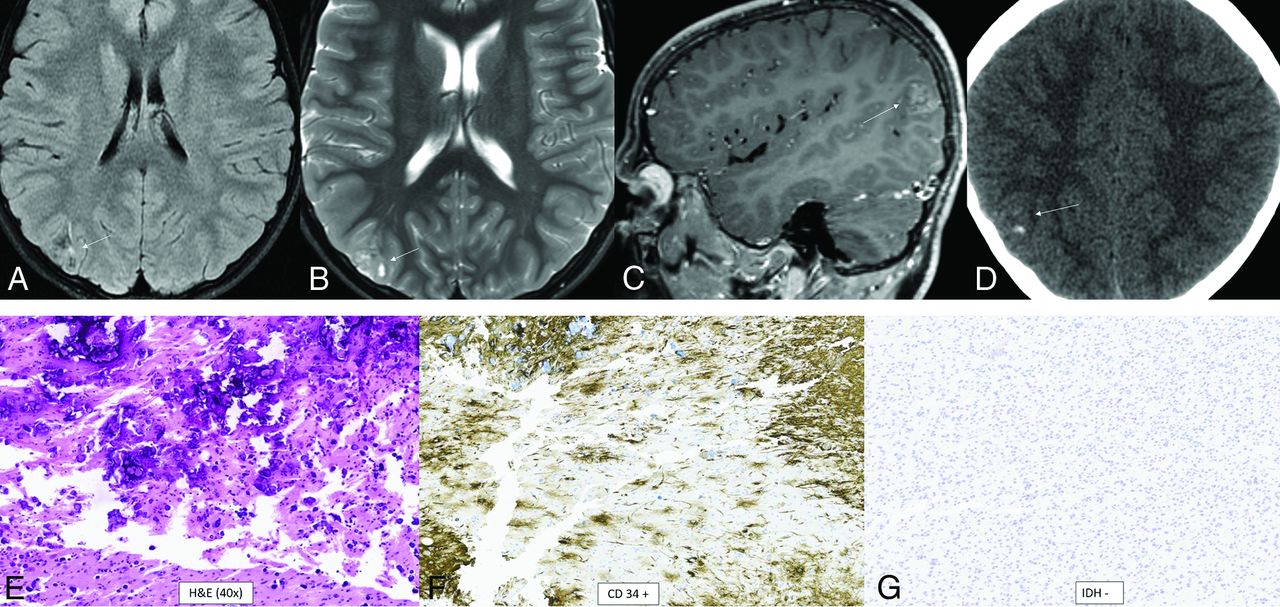
Polymorphous low-grade neuroepithelial tumor of the young is another of the 4 types of pediatric low-grade tumors and is definitionally WHO grade 1.1 It is a glial tumor with oligodendrocytic features, frequent calcifications, and an infiltrative growth pattern. It is characterized by strong CD34 immunostaining and mitogen-activated protein kinase (MAPK) pathway alterations, specifically involving FGFR or BRAF.18 The most specific alteration appears to be FGFR2-CTNNA3 fusion.16 The median age at diagnosis is 15.5 years (range, 5–57 years), with a slight female predominance (male/female ratio, 1:1.7), and epilepsy is the most common presentation (87%).19 The tumor is located supratentorially, almost always cortically or subcortically, with two-thirds in the temporal lobe. Prominent dense calcifications are classic, with calcifications occurring in 83% of cases. Typical tumors are well-circumscribed, solid, and cystic, T1- and T2-signal variable, T2-FLAIR hyperintense, and nonenhancing or mildly enhancing (Fig 2).19

Polymorphous low-grade neuroepithelial tumor of the young. It is a cortical/subcortical mass on T2-FLAIR (A) and T2-weighted images (B) with a cystic (“bubbly”) appearance, some suppression of fluid signal on the FLAIR image, and faint heterogeneous enhancement (C) within the right inferior parietal lobule (arrows). CT shows faint specks of calcification within the lesion (D). Histology demonstrates a relatively well-demarcated low-grade neuroepithelial tumor with prominent dystrophic calcification (E). Tumor cells have oligodendroglial-like morphology and are strongly positive for CD34 (F), with low proliferative activity. Immunohistochemical stain is negative for IDH1 R132H (G) and positive for OLIG2. Chromosomal microarray identified loss of 10q21.3q26.13 disrupting CTNNA3 and FGFR2, representing a FGFR2-CTNNA3 fusion.
Diffuse Low-Grade Glioma, MAPK Pathway–Altered
Diffuse low-grade glioma is another of the low-grade pediatric tumors. While not yet assigned a WHO grade, histologically, it behaves like a WHO grade 2 tumor with an oligiodendroglial, astrocytic, or mixed pattern with infiltrative growth and typical low-grade cellular features.16 Numerous molecular alterations can activate the oncogenic MAPK pathway. More common alterations involve FGFR1 and BRAF; less common alterations involve NTRK1/2/3, MET, FGFR2, and MAP2K1. IDH1/2 and H3F3s mutations and CDKN2A homozygous deletions must be absent.16 This tumor commonly presents with epilepsy in the pediatric population and occasionally in adults. While there is a paucity of literature on the radiologic findings of this tumor, given its histopathologic similarity to the other pediatric low-grade tumors, it is presumed that the imaging findings are also similar. A T2-FLAIR and T2-hyperintense, nonenhancing, cortical, temporal lobe mass is demonstrated (Fig 3).

Diffuse low-grade glioma, MAPK pathway–altered. A cortical T2-FLAIR (A) and T2-hyperintense mass (B) is noted within the right fusiform gyrus (arrows) with low T1 signal and no enhancement (C). A histologic section shows a diffusely infiltrating glioma of low cellularity with no mitotic activity, microvascular proliferation, or necrosis (D). Immunohistochemical staining is positive for GFAP (E) and shows a low Ki-67 proliferation index (F). Targeted next-generation sequencing identified a QKI-NTRK2 fusion, suggestive of MAPK pathway alteration.
Diffuse Hemispheric Glioma, H3 G34-Mutant
Diffuse hemispheric glioma is a high-grade pediatric-type tumor, definitionally WHO grade 4.1,20 Histopathologic features are similar to those of either glioblastoma or what was previously called a primitive neuroectodermal tumor. The glioblastoma-type tumors are malignant, hypercellular gliomas with astrocytic differentiation, high mitotic rates, microvascular proliferation, and necrosis. The defining molecular feature of this tumor is a missense mutation of H3F3a, which codes for histone H3, causing arginine or less commonly valine to be substituted for the normal glycine 34 (when numbered using the legacy nomenclature, which does not include the initiating methionine in the numbering).1,20 There is a strong association with ATRX and TP53 mutations. PDGFRA amplification is associated with the glioblastoma morphology. CCND2 amplification is associated with the primitive neuroectodermal tumor morphology.1,20 The median age at diagnosis is 15.8 years (interquartile range, 13–22 years), with a slight male predominance (male/female ratio, 1.5:1).21 The frontal and parietal lobes are the most common locations with frequent abutment of leptomeningeal or ependymal surfaces. Margins may be sharp or ill-defined. Most tumors are hyperdense, T1-hypointense, T2-hyperintense, enhancing, and diffusion-restricting. In adults, there may be no or only faint enhancement, in which case diffusion restriction is more helpful in assessing aggressiveness.22 Tumoral hemorrhage and necrosis can be seen and occasionally calcification (Fig 4).23

Diffuse hemispheric glioma, H3 G34-mutant. Multifocal masses are seen in the right temporal and parietal lobes. The temporal mass shows heterogeneously increased T2 signal (A), heterogeneously low T1 signal with a few foci of T1-hyperintense hemorrhage (B), and heterogenous enhancement (C, arrows). The parietal mass shows similar signal characteristics with heterogeneous T2-FLAIR hyperintensity (D), T1-hypointensity (E), and enhancement (F, arrows). The histologic section reveals an infiltrating glioma with astrocytic morphology (G). Glioma cells are positive for ATRX (H) and GFAP (I) stains and negative for IDH1 R132H and OLIG2. There was a high Ki-67 proliferation index of up to approximately 20%. This immunophenotype suggested a mutation of H3 G34, warranting further genomic evaluation. Next-generation sequencing revealed a somatic mutation in H3-3A (also known as H3F3A). Currently, there are no clinically approved therapies specifically targeting H3-3A mutations.
Diffuse Pediatric-Type High-Grade Glioma, H3 Wild-Type and IDH Wild-Type
Diffuse pediatric-type high-grade glioma is another of the 4 pediatric high-grade glioma types. It does not have an assigned WHO grade or a single defining molecular or genetic feature.1 About half of tumors previously classified as “pediatric glioblastoma” demonstrate mutations of histone 3 or uncommonly IDH1/2. The remaining heterogeneous tumors now fall under this new classification. The 3 recognized subtypes are characterized by MYCN, PDGFRA, and EGFR amplifications with numerous coexisting genetic abnormalities described.24 The MYCN subtype has high cellularity and mitosis, spindle, and epithelioid cell components; necrosis; and microvascular proliferation.25,26 The median age at diagnosis is 8–11 years (range, 2–18 years).24 There is no sex predilection overall, but there is a slight male predominance for the EGFR subtype (male/female ratio, 1.6:1).24 The location is usually supratentorial, with the posterior fossa approaching 20% of cases, depending on subtype.24 The MYCN subtype classically shows a solid, enhancing, diffusion-restricting, well-marginated temporal lobe mass abutting the meninges with tumoral necrosis, rare hemorrhage, and no calcifications.25,26 A tumor in the pons has greater enhancement and diffusion restriction compared with a diffuse midline glioma, H3 K27-altered.25 Figure 5 demonstrates a less-typical case without enhancement of the primary tumors.

Diffuse pediatric-type high-grade glioma, H3 wild-type and IDH wild-type. MR images demonstrate diffusely infiltrating masses in the bilateral temporal lobes with mild mass effect, T2-FLAIR (A) and T2-hyperintensity (B, anterior arrows), and no enhancement (C). There is additional T2-FLAIR and T2-hyperintensity in the dorsal pons and posterior fossa leptomeningeal enhancement (posterior arrows). Histology reveals a high-grade diffusely infiltrating astrocytoma with high mitotic activity (D). The tumor shows ATRX retention (E) and wild-type IDH status (F) on immunohistochemical stains. Whole-genome methylation analysis showed a match to diffuse pediatric-type high-grade glioma. The tumor was H3 wild-type and IDH wild-type and had a TERT promoter mutation, which was identified on the neuro-oncology targeted next-generation sequencing panel.
Infant-Type Hemispheric Glioma
Infant-type hemispheric glioma is a pediatric-type, diffuse, high-grade glioma that has not yet been assigned a specific WHO grade. The hallmark of this tumor is receptor tyrosine kinase gene fusions of ALK, ROS1, NTRK1/2/3, or MET.1 NTRK3 fusion has also been described in congenital mesoblastic nephroma and congenital fibrosarcoma, implying that such genetic alterations are tied to age-related mechanisms.27 Most of these tumors show high-grade histologic features.27 Histopathology shows hypercellularity, astrocytic differentiation, necrosis, microvascular proliferation, and nuclear pleomorphism.28 The median age at diagnosis is 2.8 months (range, 0.0–12.0 months) with no sex predilection. Overall median survival is 1.9 years.27 The tumors are almost always located in the cerebral hemispheres.27 Imaging data are scarce, but tumors tend to be large with solid and prominent cystic components, intratumoral hemorrhage, and enhancement.29⇓-31 Leptomeningeal disease has been reported.30
High-Grade Astrocytoma with Piloid Features
High-grade astrocytoma with piloid features is a circumscribed astrocytic glioma that has not yet been assigned a WHO grade but behaves like WHO grade 3 or 4.8,15 A hallmark of this tumor is its unique methylation profile.32 The most common genetic abnormalities are cdkn2A/B deletion, MAPK pathway alteration (affecting NF1, BRAF, and FGFR1), and ATRX mutation or loss of expression. Histologically, tumors tend to show moderate cellularity, glioblastoma-like foci, moderate nuclear pleomorphism, a moderate mitotic rate, lack of necrosis, vascular hypertrophy, and infiltrative growth.32 Most occur in the posterior fossa (74%), usually in the cerebellum, followed by supratentorial then spinal locations. The median age is 41.5 years, with occurrence from pediatrics to the elderly and no sex predilection.32 There appears to be an association with neurofibromatosis type 1.33 Tumors tend to be T1-hypointense-to-isointense, T2-hyperintense, heterogeneously enhancing, non-diffusion-restricting, and non-necrotic with sharp or ill-defined margins (Fig 6).12

High-grade astrocytoma with piloid features. MR images demonstrate an eccentric T2-hyperintense mass (A) along the posterior thoracic cord with low T1 signal (B) and avid enhancement (C–E, arrows). Surrounding intramedullary T2-hyperintensity represents edema and/or tumor infiltration. Histology reveals an astrocytoma with piloid morphology (F), having NF1 (G) and ATRX (H) mutations in association with CDK2NA/B homozygous deletion. Whole-genome methylation profiling showed a match to high-grade astrocytoma with piloid features. Most of these tumors occur intracranially.
Diffuse Glioneuronal Tumor with Oligodendroglioma-Like Features and Nuclear Clusters (Provisional Type)
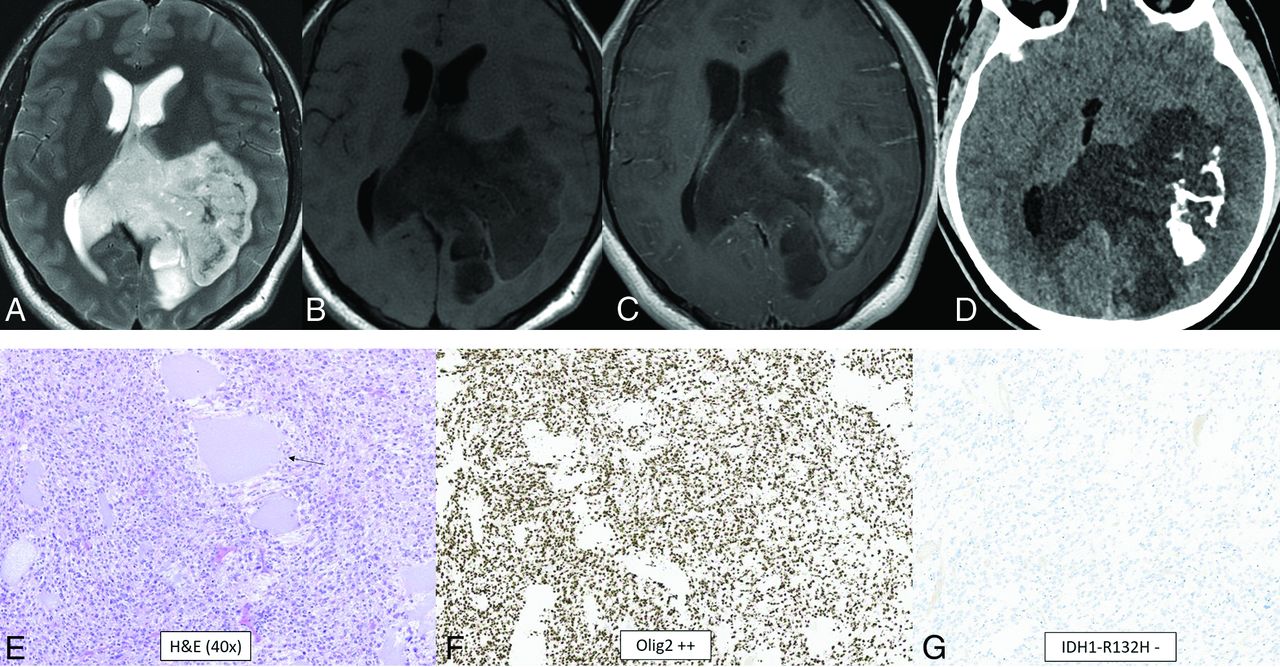
Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters is a provisional tumor that has not yet been assigned a WHO grade. The hallmark of this tumor is its unique methylation profile.34 Additionally, monosomy 14 is seen in almost all cases. Histologically, the tumors tend to have oligodendroglioma-like perinuclear haloes, clear cell morphology, vascular growth, nuclear clusters resembling “pennies on a plate,” moderate-to-high cellularity, and infiltrative growth.34 Calcifications have been reported.35 The median age is 9 years (range, 2–75 years), and there is no sex predilection.34 Location is usually in the cerebral hemispheres, more commonly in the temporal lobe.34 A typical tumor is solid and cystic, T1-hypointense, T2-hyperintense, and nonenhancing-to-minimally enhancing with calcifications and without adjacent edema (Fig 7).35

Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (provisional type). CT and MR images demonstrate a large well-circumscribed mass centered at the left atrium with involvement of the left parietal lobe and transcallosal extension to the right hemisphere. The tumor shows T2-hyperintensity (A), T1-hypointensity (B), patchy heterogeneous enhancement (C), and dense calcification (D). The histologic section demonstrates a highly cellular infiltrating glioma with oligodendroglioma-like microcyst formation (arrow, E) and extensive calcification. The tumor cells show moderate, clear cytoplasm and round-to-elongated, irregular, and hyperchromatic nuclei with high mitotic activity. The tumor cells are diffusely positive for OLIG2 (F) and negative for IDH1 R132H (G) and H3 K27M by immunohistochemistry. Whole-genome methylation analysis confirmed the final integrated diagnosis of diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters.
Myxoid Glioneuronal Tumor
Myxoid glioneuronal tumor is a benign WHO grade 1 tumor that shows low-grade oligodendrocyte-like tumor cells with a myxoid-/mucin-rich stroma on histology. A fine capillary network is sometimes present along with neurocytic rosettes. Glial fibrillary acidic protein (GFAP) and OLIG2 are positive. The defining feature is a PDGFRA p.K385 mutation. Abnormalities in FGFR1, IDH1/2, BRAF, MYB, and MYBL1 are absent.36,37 Data are limited, but in the largest described series, the median patient age was 23.6 years (range, 6–65 years) with no sex predilection.38 These tumors have a propensity for the septum pellucidum.36,38 A typical mass is well-defined, lobulated, T1-hypointense, T2-hyperintense, nonenhancing, non-diffusion-restricting, and without surrounding edema. T2-FLAIR shows relative hypointensity centrally and hyperintensity peripherally (Fig 8). There is no elevated CBF. Larger lesions can appear L-shaped and have mass effect, which can mimic high-grade tumors.36⇓⇓-39

Myxoid glioneuronal tumor. MR imaging shows a mass in the right frontal horn region with peripheral T2-FLAIR hyperintensity and relative central T2-FLAIR hypointensity (A), which is slightly hypointense to CSF on the T2-weighted image (B), approaches the septum pellucidum (arrow, B), and does not enhance (C). There is no adjacent edema. Histology shows an oligodendroglioma-like appearance with moderate cellularity and uniform round nuclei with a circumferential arrangement around delicate vessels (D). Immunohistochemical staining is positive for OLIG2 (E), synaptophysin (F), and GFAP and negative for NeuN. This tumor has a PDGFRA p.K385 mutation and lacks FGFR1 abnormalities. Dysembryoplastic neuroepithelial tumor is a histologic mimic but has a mutation of FGFR1.
Multinodular and Vacuolating Neuronal Tumor
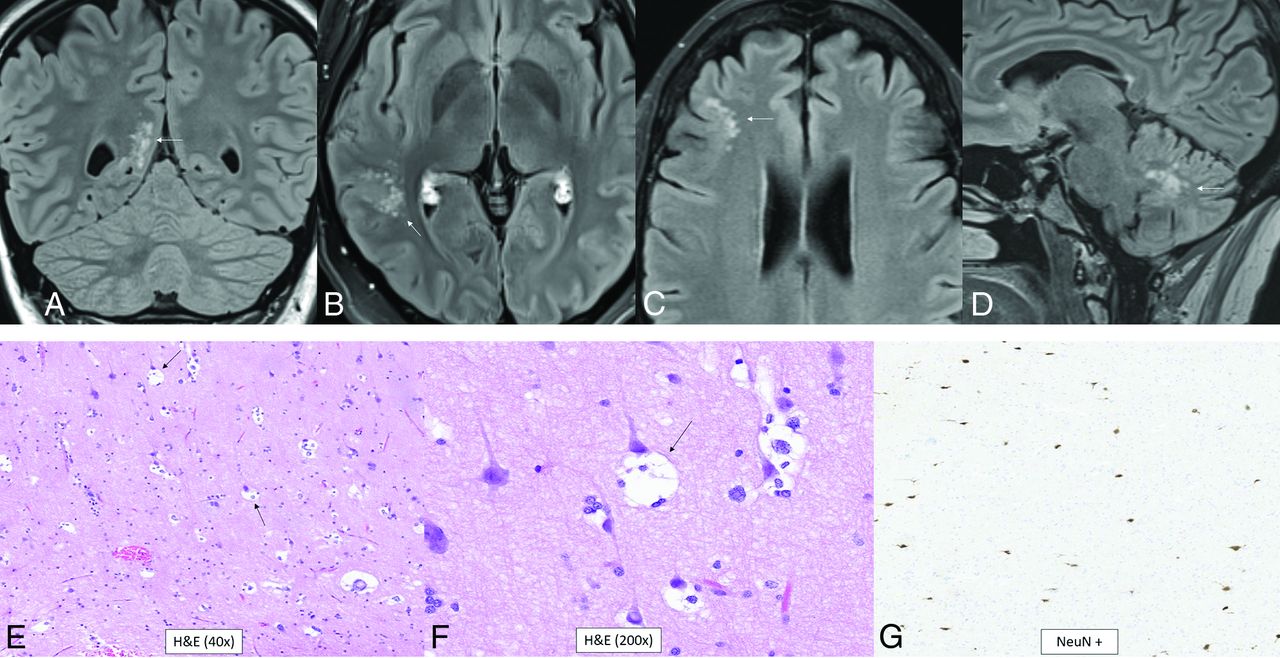
Most of the multinodular and vacuolating neuronal tumors are WHO grade 1 tumors that have a MAP2K1 mutation, but FGFR2-ZMYND11 translocations and BRAF, DEPDC5, SMO, TP53, PIK3CA, and CIC mutations can also occur.16,40 Histologically, there are multiple, discrete, and coalescent nodules with immature neuronal cells and round vesicular nuclei. Pericellular eccentric vacuolization with prominent nucleoli and eosinophilic cytoplasm are seen. There is no mitosis, perivascular lymphocytic infiltration, microcalcification, or oligodendroglia-like cells. OLIG2, α-internexin, and synaptophysin are positive.40 The median age is 41 years (range, 8–63 years), and there is a slight female predominance (male/female ratio, 1:1.4).41 The tumor presents as cluster of variably-sized nodules in the subcortical ribbon and superficial subcortical white matter following the gyral contour. The frontal then parietal, occipital, and temporal lobes are the most common locations.41,42 The tumors are T1 iso- to hypointense, T2-hyperintense, nonenhancing, non-diffusion-restricting, and without mass effect, calcification, hemorrhage, or surrounding edema (Fig 9).41⇓⇓-44

Multinodular and vacuolating neuronal tumor. Four different patients with pathology-proved (A, and E–G) and radiologic (B–D) diagnoses. T2-FLAIR images show small, subcortical clusters of hyperintense nodules with no edema or mass effect (A–D, arrows). Histology shows “neuronal” tumor cells with eosinophilic cytoplasm (E), multiple intracytoplasmic vacuoles that markedly enlarge the cytoplasm (arrows, E and F), and perineuronal satellitosis. There is positive NeuN staining of the viable normal scattered pyramidal neurons with negative staining of the tumor cells (G).
Supratentorial Ependymoma, YAP1 Fusion-Positive
Supratentorial ependymomas, which are WHO grade 2 or 3, are associated with many different mutations with YAP1 fusions accounting for only 7% of all supratentorial ependymomas.30 Within this group, YAP1-MAMLD1, and YAP1-FAM11B fusions are common.45⇓⇓-48 Histologically, bipolar spindle cells with elongated processes are seen among blood vessels. Perivascular anuclear zones form perivascular pseudorosettes, and some cells have cytoplasmic vacuolization. Neoplastic nuclei are moderate in size and round-to-ovoid with speckled chromatin. Rosenthal fibers are seen in the zone surrounding the tumor.46,47 Irregular cells resembling tancytes are visualized. GFAP, S-100, and vimentin are positive.45⇓-47 Data are limited, but the reported median age is 1.4 years (range, 0–51 years) with most patients younger than 4 years of age and almost all younger than 9 years of age. There is a female predominance (male/female ratio, 1:3).45 Location is within the lateral ventricles or in the brain parenchyma adjacent to them.30 For extraventricular, supratentorial ependymomas in general, the frontal and temporal lobes are the most common locations.46 A typical tumor is mixed density, solid and cystic, well-marginated, T1 iso- to hypointense, T2 iso- to hyperintense, enhancing, and diffusion-restricting with calcifications. Internal hemorrhage can occur.46,48,49
Posterior Fossa Ependymoma, Group PFA
Posterior fossa ependymomas are divided into groups PFA and PFB. PFA is a WHO grade 2 or 3 tumor characterized by loss of H3 K27 trimethylation due to EZHIP overexpression.50⇓-52 PFA tumors are further divided into PFA-1 and PFA-2 based on the specific mutation present. PFA-1 has HOX mutations while PFA-2 has EN2 and CNPY1 mutations.9,50⇓-52 Histology demonstrates well-differentiated cells with ependymal rosettes and perivascular pseudorosettes. Dystrophic calcification, hemorrhage, myxoid degeneration, and metaplasia can also be seen. GFAP and S-100 are positive, and OLIG2 is negative.53 The median age is 3 years (range, 0–51 years) with most patients younger than 9 years of age.45 There is a slight male predominance (male/female ratio, 1.8:1). Overall survival at 5 and 10 years of age is 68% and 56%, respectively.45 PFAs account for nearly 90% of all posterior fossa ependymomas.30 Tumors arise from the roof of the fourth ventricle or the cerebellopontine cistern, can traverse the foramina of Luschka or Magendie, and can encase cranial nerves and vessels.52,53 The typical imaging appearance shows calcification, cystic change, T1 iso- to hypointensity, T2-hyperintensity, and heterogeneous enhancement. Hemorrhage and diffusion restriction can be present.52,53
Posterior Fossa Ependymoma, Group PFB
PFB is a WHO grade 2 or 3 tumor characterized by increased H3 K27 trimethylation.50⇓-52 PFB ependymomas also arise from the fourth ventricle but more commonly from the floor as opposed to the roof. In comparison with PFA, PFB tumors more commonly occur in adolescents and young adults.30,54 On the basis of supplemental data from the largest reported series, the overall median age is 27.5 years (range, 1–72 years) with no sex predilection, though there are age and sex differences among PFB subtypes.55 Prognosis is substantially better than for PFA, with overall survival at 5 and 10 years being 100% and 88%, respectively.45 The histology and immunohistochemistry findings are similar to those of PFA. The imaging findings are also similar; however, compared with PFA tumors, PFB tumors tended to be more cystic, less calcified, and less enhancing.54
Spinal Ependymoma, MYCN-Amplified
Spinal ependymoma is a rare, aggressive tumor of the spinal cord.56,57 While not yet assigned a specific WHO grade, its histologic features are usually WHO grade 3 but can be WHO grade 2.56,58 Its defining feature is amplification of MYCN, which has been implicated as the driver of its aggressive behavior.56 Histologically, this tumor is anaplastic with marked cellular atypia and nuclear hyperchromasia. Prominent pink nucleoli, necrosis, mitosis, and glomeruloid vascular proliferation are common. GFAP and EMA are positive.56,58 While data are limited, the reported median age is 32 years (range, 12–56 years) with no sex predilection.58 These tumors can grow to be large and cause spinal canal widening. A typical tumor is well-demarcated, iso- to hyperdense, T1 iso- to hypointense, and T2 iso- to hyperintense. Enhancement is variable. A hemosiderin rim (“cap sign”) may form from tumoral hemorrhage.30,56,59,60
Cribriform Neuroepithelial Tumor (Provisional Type)
Cribriform neuroepithelial tumor is a benign tumor that has not yet been assigned a WHO grade. It is defined by a large, heterozygous deletion in SMARCB1, which is also seen in atypical teratoid/rhabdoid tumor (AT/RT).30,61,62 The key histologic features of cribriform neuroepithelial tumor are the presence of cribriform strands, ribbons, and nuclei with dense chromatin. Cells lack prominent nucleoli, and the cytoplasm is slightly eosinophilic and ill-defined. In more compact areas, small lumina may be seen with true rosettes.61,63,64 Tyrosinase, EMA, vimentin, MAP2C, and synaptophysin are positive.61,65,66 While data are limited, the reported median age is 1.7 years (range, 0.8–10.8 years) without a definite sex predilection.65 The location is intraventricular or within the brain parenchyma adjacent to the ventricles. Tumors have been described in the lateral, third, and fourth ventricular regions without a clear predilection for 1 of these 3 locations.65 Imaging typically reveals a large mass with T1-hypointensity, T2-hyperintensity, heterogeneous enhancement, and diffusion restriction.
CNS Neuroblastoma, FOXR2-Activated
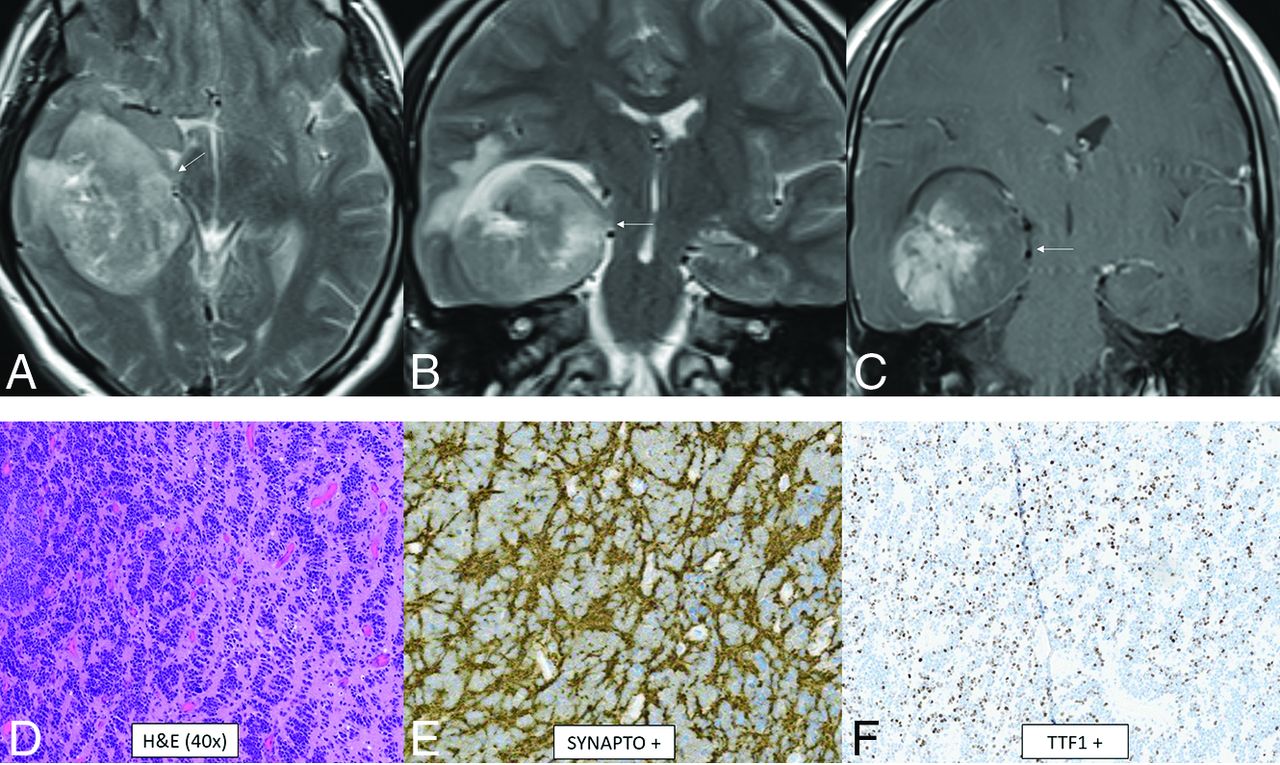
CNS neuroblastoma is a highly malignant embryonal tumor without an official WHO grade. These tumors have variable chromosomal rearrangements or mitochondrial DNA insertions converging on FOXR2, leading to overexpression.67,68 FOXR2 binds to and stabilizes MYC and MYCN proteins and therefore promotes MYC-related transcriptional activities, leading to increased cellular proliferation and tumorigenesis.67,68 Histology shows a small-cell tumor, embryonal architecture, a high proportion of neuropil, neurocytic cell, or ganglion cell differentiation, and, frequently, vascular pseudorosettes and nuclear palisades.67 OLIG2 and synaptophysin are positive.67 The median age is 4.5 years (range, 1.4–16 years) without a sex predilection.69 In the largest described series, tumors were large and supratentorial with invariable involvement of the deep white matter and frequent invasion of the cortex (80%) as well as a ventricular ependymal surface (64%).69 The frontal lobe was the most common location, though involvement of multiple regions was common. Typical tumors were multilobulated, solid, and cystic/necrotic, T2-hyperintense, enhancing, and diffusion-restricting (Fig 10). Calcification or hemorrhage was present in approximately 40% of tumors. Calvarial remodeling was present in nearly half of cases and occurred more frequently with larger tumors.

CNS neuroblastoma, FOXR2-activated. MR images show a large cortical and subcortical well-circumscribed expansile mass in the right temporal lobe involving the hippocampus (arrows) with T2-hyperintensity (A and B) and patchy enhancement (C). Histology shows a highly cellular infiltrating neuroepithelial neoplasm with a complex pattern, including an undifferentiated and extensive spongioblastoma pattern with brisk mitotic activity (D). On immunohistochemistry, the tumor shows divergent differentiation with expression of synaptophysin (E), OLIG2, and TTF1 (F). Whole-genome methylation profiling indicated a match to CNS neuroblastoma, FOXR2-activated.
CNS Tumor with BCOR Internal Tandem Duplication
CNS tumor with BCOR internal tandem duplication is a high-grade tumor that has not yet been assigned a specific WHO grade. Histologically, it is a compact tumor containing spindle and oval cells with fine chromatin with a well-demarcated border with adjacent brain parenchyma.67,70 Perivascular pseudorosettes with an ependymoma-like appearance and an intervening anuclear zone are prominent.67 Peripheral calcified palisading necrosis is typically present. Tumors often demonstrate fibrillary processes and contain dense capillary networks.67,70 GFAP and S-100 are negative, and BCOR, OLIG2, and NeuN are positive.67 Data are limited, but the reported median age is 1.8 years (range, 1.2–7.6 years) with a female predominance (male/female ratio, 1:2.3).70 Location can be supratentorial or infratentorial, but dural abutment is common. A typical tumor is large, solid, centrally necrotic, iso- to hypodense, T2-hyperintense, diffusion-restricting, and mildly enhancing. Calcification or blood products are sometimes present at the border of the necrotic region. Large intratumoral macroscopic vessels may be present (Fig 11).70

CNS tumor with BCOR internal tandem duplication. In 1 patient (A–C), there is a large well-circumscribed mass in the right posterior frontal lobe with heterogeneous T2-hyperintensity and prominent intratumoral vessels (A), heterogeneous T1-hypointensity (B), and enhancement (C, arrows). In a different patient (D–F), there is similar signal and morphology of a mass centered in the left cerebellar hemisphere on the T2-weighted (D), T1-weighted (E), and T1-weighted postcontrast (F) images (arrows). Histology from the first patient is characterized by relatively uniform nuclei, perivascular arrangement of tumor cells (rosette formation), and necrosis without microvascular proliferation (G). Immunohistochemical stains demonstrate OLIG2 positivity (H) and consistent NeuN positivity (I). Next-generation sequencing identified a frameshift mutation in BCOR, and chromosomal microarray demonstrated a segmental chromosomal loss disrupting BCOR.
Desmoplastic Myxoid Tumor of the Pineal Region, SMARCB1-Mutant
Desmoplastic myxoid tumor is a tumor of the pineal region without a specific WHO grade that has a mutation in SMARCB1, resulting in a loss of function, similar to AT/RT.71,72 Histologically, there is no brisk mitotic activity or necrosis, typically seen in AT/RT. These tumors have a variable myxoid morphology combined with spindled and epithelioid cells embedded within a densely collagenized stroma. CD34 is positive, and INI1 is negative.71⇓-73 Unlike AT/RT, this tumor more commonly occurs in adults (median age, 40 years; range, 15–61 years).15 Data are limited, but there does not appear to be a sex predilection.72 On imaging, there is variable T1 signal, T2 intermediate signal, and enhancement.71,73 Large tumors can compress the cerebral aqueduct and cause obstructive hydrocephalus.71
Intracranial Mesenchymal Tumor, FET-CREB Fusion-Positive (Provisional Type)
Intracranial mesenchymal tumor is a group of rare mesenchymal CNS tumors without an assigned WHO grade. Intracranial angiomatoid fibrous histiocytomas and intracranial myxoid mesenchymal tumors have now been combined into this group because both have an in-frame genetic fusion of a FET RNA-binding protein (EWSR1 or FUS) to a CREB transcription factor (ATF1, CREB1, or CREM).74 Histology is variable and may show solid nodules of epithelioid or spindled cells with a syncytial growth pattern, pseudoangiomatous spaces, a fibrous pseudocapsule, prominent pericapsular lymphoplasmacytic infiltrates, or mucin-rich stroma.74 Desmin, CD99, and EMA are positive, and skeletal and smooth muscle markers, S-100, GFAP, and OLIG2 are negative.74 The median age is 17 years (range, 4–70 years) with a female predominance (male/female ratio, 1:2.2).74 Location is typically extra-axial, most commonly along the cerebral convexities but can be intraventricular or infratentorial.15 A dural tail and calvarial involvement may be present. A typical tumor is well-circumscribed, lobulated, and cystic and solid with T2 and T2-FLAIR hyperintensity and enhancement. Internal blood products may be present. Extensive adjacent vasogenic edema is common (Fig 12).15

Intracranial mesenchymal tumor, FET-CREB fusion–positive (provisional type). Sagittal (A–C) and axial (D–F) MR images demonstrate a lobulated circumscribed mass along the superior vermis. The mass is markedly T2-FLAIR hypointense (A and D), T1-hypointense (B), homogeneously enhancing (C), and T2-hyperintense (E), with marked surrounding vasogenic edema. There is a lack of hypointensity within the tumor on the susceptibility-weighted image (F). Histology demonstrates a mesenchymal neoplasm with low-grade features (G), with staining positive for reticulin (H) and desmin (I), markers of connective tissue and muscle, respectively. The marked T2-FLAIR hypointensity corresponding to the area of homogeneous enhancement is an atypical appearance.
CIC-Rearranged Sarcoma
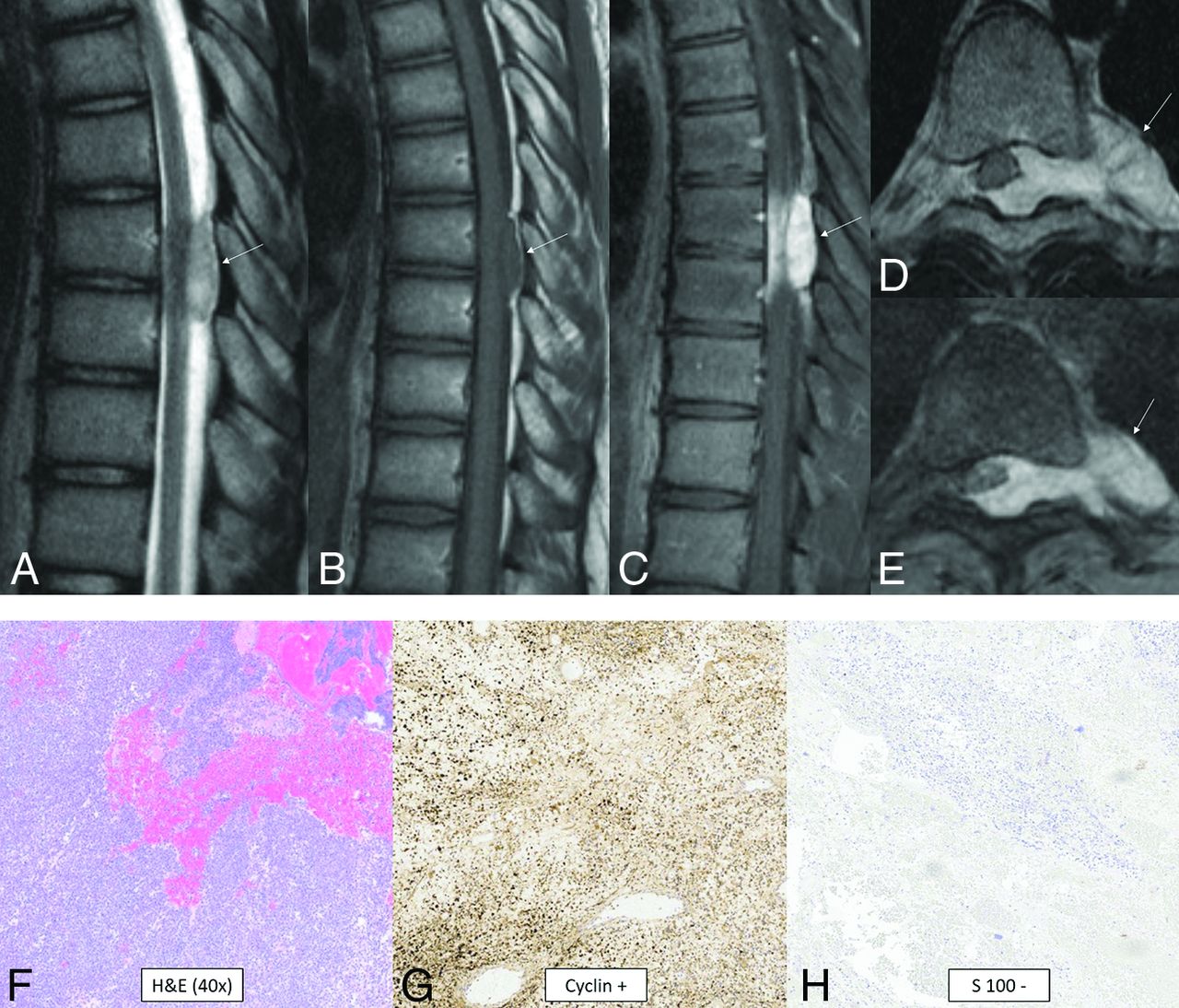
CIC-rearranged sarcoma is a highly aggressive WHO grade 4 round cell mesenchymal neoplasm that is one of the most common and best characterized subgroups of “Ewing-like sarcomas”75 and is predominantly extraskeletal.76 These tumors are characterized by CIC rearrangements with multiple fusion partners identified (DUX4, FOXO4, LEUTX, NUTM1, NUTM2A).75,76 The CIC-NUTM1 fusion pair appears to have a greater predilection for the CNS.67,75 Histologically, they are small, round cell tumors, but in contrast to Ewing sarcoma, they exhibit distinctive nucleoli in cells with vesicular nuclei, variable epithelioid morphology occasionally with clear cytoplasm, focal myxoid change and cell spindling, and reduced uniformity of nuclei size and shape.75,76 CD99, ETV4, and WT1 are positive.75,76 Data are limited for CIC-rearranged sarcoma of the CNS, but cases have been reported in both pediatric and adult patients.77,78 Location is anywhere along the neuroaxis.79,80 A typical tumor is extra-axial, solid, variably lobulated; T2 iso- to hyperintense; and homogeneously or heterogeneously enhancing (Fig 13). Peritumoral edema can be present.81

CIC-rearranged sarcoma. MR images reveal a small well-circumscribed peridural mass in the midthoracic region (arrows). The mass is T2-hyperintense (A), T1-isointense (B), and avidly enhances (C–E). The tumor extends through the left neural foramen into the paraspinal space and also exerts moderate mass effect on the cord, displacing it to the right (D and E). Histology reveals small, round, sarcomatous cells (F) with molecular stains of neoplastic cells positive for cyclin (G), calretinin, WT1 (focal), and CD99 (patchy) and negative for S-100 (H), CK, EMA, desmin, and synaptophysin. Fluorescence in site hybridization showed a balanced rearrangement of the CIC locus in 91% (181/200) of the nuclei.
Primary Intracranial Sarcoma, DICER1-Mutant
Primary intracranial sarcoma is a highly malignant tumor, associated with familial DICER1 syndrome and occasionally neurofibromatosis type 1.15,82,83 A specific WHO grade has not yet been assigned. There are several other DICER1-associated tumors in and outside the CNS.82 DICER1 encodes a protein that facilitates activation of an RNA-induced silencing complex. Disruption of this pathway leads to altered protein expression, activation of the NRAS variants, inactivation of TP53, and copy number alternations.82 Histologically, there is high cellularity, brisk mitotic activity, intratumoral hemorrhage, some areas of fascicular spindle cells, and embryonic-type tissues, which may have rhabdomyoblastic differentiation.82,83 Coalescence of cells into “organoid” formations has been observed.82 PAS, α-1 antitrypsin, and desmin are positive, with patchy-to-complete loss of H3K27me.82,83 The median age is 6.0 years (range, 2.0–17.5 years) without a sex predilection.84 Tumors are presumed to arise from mesenchymal progenitor cells located within the meninges or perivascular spaces83 and thus can present as intra-axial or extra-axial masses. Intra-axial masses tend to be peripheral and within the cerebral hemispheres.82⇓⇓⇓-86 The typical appearance is hyperdense, T2 iso- to hypointense, diffusion-restricting, and enhancing, with intratumoral hemorrhage and peritumoral edema.82⇓⇓-85,87
Pituitary Blastoma
Pituitary blastomas are rare WHO grade 4 embryonal tumors of the adenohypophysis associated with DICER1 mutations.15,85,88 Histologically, these tumors resemble the embryonic pituitary gland and are composed of blastema-like cells, epithelial glands with rosettes resembling primitive Rathke-type epithelia, and large secretory epithelial cells that express hormones such as adrenocorticotropic hormone or rarely growth hormone.85,89 In the largest reported series, the median age was 11 months (range, 2–24 months) with a slight female predilection (male/female ratio, 1:1.4),90 though a case in a young adult has been described.89 The most common clinical presentation is an infant with Cushing syndrome, ophthalmoplegia, and/or diabetes insipidus.85 The imaging appearance is variable, ranging from a small pituitary mass to a large heterogeneous solid and cystic mass mimicking a macroadenoma.89 Internal calcification has been reported in 1 case.89
CONCLUSIONS
The 2021 version of the WHO CNS tumor classification includes terminology updates reflecting more accurate understanding of tumorigenesis as well as the presentation of 22 newly recognized tumors, which were reviewed here. This edition furthers the growing movement away from purely histologic diagnoses and toward molecular diagnoses, increasing the emphasis on specific genetic mutations and DNA methylation–based classification. Because this classification system improves standardization in diagnosis and facilitates targeted treatments, it will continue to grow and adapt on the basis of new understanding of molecular alterations and tumor pathogenesis.
Footnotes
Disclosure forms provided by the authors are available with the full text and PDF of this article at www.ajnr.org.
Indicates open access to non-subscribers at www.ajnr.org
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- Received November 2, 2022.
- Accepted after revision December 14, 2022.
- © 2023 by American Journal of Neuroradiology




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Mesenchymal Nonmeningothelial Tumors of the CNS: Evolving Molecular Landscape and Implications for Neuroradiologists
- Imaging Features of Primary Intracranial Sarcoma with DICER1 Mutation: A Multicenter Case Series
- High-Grade Astrocytoma with Piloid Features: A Dual Institutional Review of Imaging Findings of a Novel Entity