
Abstract
SUMMARY: Alzheimer disease affects millions of people worldwide. The neuropathologic process underlying this disease begins years, if not decades, before the onset of memory decline. Recent advances in neuroimaging suggest that it is now possible to detect Alzheimer-associated neuropathologic changes well before dementia onset. Here, we evaluate the role of recently developed in vivo biomarkers in the clinical evaluation of Alzheimer disease. We discuss how assessment strategies might incorporate neuroimaging markers to better inform patients, families, and clinicians when memory impairment prompts a search for diagnosis and management options.
ABBREVIATIONS:
- AD
- Alzheimer disease
- APOE ε4
- apolipoprotein ε4
- FTD
- frontotemporal dementia
- MCI
- mild cognitive impairment
- NFTs
- neurofibrillary tangles
- PIB
- Pittsburgh Compound-B
- vMRI
- volume-based MR imaging; amyloid-β Aβ
Late-onset Alzheimer disease (AD) is the most common form of dementia with an estimated prevalence of 30 million people worldwide, a number that is expected to quadruple in 40 years. With increasing awareness that symptoms develop over many years, there is a growing need to identify nondemented older people at risk for AD. Mild cognitive impairment (MCI) represents a transitional state between normal aging and dementia. Clinical features of amnestic MCI are presented in Table 1 and are reviewed by Petersen et al1 and again by Petersen.2 In this piece, we focus on recent advances in biomarker development for the predictive prognosis of MCI and suggest that a neuroimaging-based evaluation strategy can help guide clinical management decisions in older people with memory impairment.
Clinical features in amnestic patients with MCI
AD Pathobiology
Since their first description by Alois Alzheimer in 1907,3 amyloid-containing plaques and tau-associated neurofibrillary tangles (NFTs) have remained the 2 hallmark pathologic lesions of AD. Senile and neuritic plaques are composed of amyloid-beta (Aβ), a 38–43 amino acid peptide that derives from the much larger cell membrane–associated amyloid precursor protein and gradually accumulates over time in the extracellular spaces of the brain.4 Within plaques, Aβ is present in aggregated/insoluble forms such as fibrils and soluble forms such as oligomers.5 In animal models, Aβ initiates downstream loss of dendrites and synapses5 and functional disruption of neuronal networks.6 Genetic evidence indicates that apolipoprotein ε4 (APOE ε4), the most important known genetic risk factor for late-onset AD, accelerates the onset of Aβ deposition into plaques and decreases the transport of Aβ across the blood-brain barrier.7 Furthermore, a recently discovered mutation in Aβ-precursor protein protects against AD,8 providing additional evidence regarding the central role of Aβ in AD pathogenesis. However, neocortical Aβ plaques are present not only in cognitively impaired patients but also in cognitively normal older adults.9 Poor correlations between Aβ deposition and memory decline,10 together with the observation that immunotherapy-induced Aβ plaque removal may not prevent neurodegeneration,11 suggest that additional entities besides Aβ are required for AD-associated degeneration.
NFTs, primarily found in neuronal cell bodies, are composed of the hyperphosphorylated, aggregated form of the microtubule-binding protein, tau. Unlike Aβ plaques, tau-associated NFTs correlate strongly with clinical severity10 and follow a defined temporal topographic pattern in which medial temporal lobe regions underlying memory function are affected in the earliest stages of the disease.12 Recent work in animal models13,14 and in humans15,16 points to a synergistic relationship between Aβ and tau whereby Aβ-associated neurodegeneration occurs only in the presence of tau. Intriguingly, evidence from animal models indicates that reducing tau levels rescues mice from premature mortality and memory deficits without altering Aβ levels or plaque burden.13 These findings, along with other biochemical and experimental evidence, support a 2-stage disease process where Aβ deposition initiates the neurodegenerative cascade (including tau hyperphosphorylation and aggregation), which in turn becomes increasingly independent of the initiating Aβ.17
Imaging and Fluid Biomarkers for Assessment of AD Pathology
Neuropathologic findings indicate that Aβ accumulation and tau pathology begins years or even decades before the onset of clinical symptoms.18 Neuroimaging and CSF markers can detect the earliest pathology associated with AD, enabling identification of clinically normal patients in the presymptomatic or preclinical stage of AD.19 In the sections below, we review the most extensively validated in vivo biomarkers of amyloid pathology and AD-related neurodegeneration. For simplicity, we do not review the putative markers of synaptic injury, such as FDG-PET or functional MR imaging, which may prove useful in distinguishing among certain neurodegenerative disorders.
Volumetric Structural MR Imaging
Structural MR imaging is a convenient first imaging technique to assess AD neurodegeneration because current practice guidelines include its use during the routine evaluation of patients with cognitive complaints, primarily to exclude structural abnormalities such as infarction, brain tumors, or hydrocephalus.20 Brain atrophy on structural MR imaging reflects the loss of dendrites, synapses, and neurons.21 Although atrophy is not specific to AD, a strong association exists between the severity of atrophy and cognitive decline along the aging continuum, and the degree of atrophy correlates with Braak pathologic staging at autopsy.21 It is important to note that the topographic distribution of MR imaging–based atrophy in AD maps well onto the distribution of NFT pathologic features, with the entorhinal cortex and hippocampus demonstrating the largest magnitude of gray matter loss in patients with a high tau burden.22
A number of methodologies, ranging from whole-brain or voxel-based techniques to region-of-interest–based methods, have been proposed to quantitatively evaluate brain atrophy on MR imaging. Within the last decade, the routine acquisition of high-quality 3D T1-weighted images and rapid advances in image analysis algorithms have led to the availability of volumetric MR imaging–based (vMRI) software tools capable of automatically subdividing the brain into neuroanatomic regions and quantifying tissue loss within each region for a single patient.23⇓–25 Fully automated quantitative vMRI-based neuroanatomic assessments can detect AD-associated volume loss, predict disease progression, and be used as an outcome measure in therapeutic trials.26,27 Recently, the FDA has approved one such vMRI technology28 that can assist in the clinical work-up of memory decline (Fig 1). In the On-line Table, we review recent (from 2009–2012) prospective studies using vMRI methods to predict clinical progression from MCI to AD.

Brain MR imaging evaluation of a patient with amnestic MCI by use of a volumetric technique (NeuroQuant, http://www.cortechslabs.com). The top panel illustrates subcortical regions, such as the hippocampus (dark yellow), automatically classified on axial, coronal, and sagittal T1-weighted MR images. The middle and bottom panel demonstrate volumes and normative percentiles for the hippocampus and ventricles. Analyses of the baseline MR imaging scan demonstrated hippocampal volumes that were at the < 1 normative percentile, lending objective support to an impression of medial temporal lobe atrophy. At the time of volumetric assessment, the patient's Mini-Mental Status Examination score was 29 of 30, yet memory impairment was suggested by more detailed neuropsychological testing. Three years later, his Mini-Mental Status Examination score was 22 of 30, and he had clinically progressed to dementia with high biomarker probability of AD, as supported by evidence of neuronal injury on structural MR imaging and elevated amyloid levels on a florbetapir scan (Fig 3).
However, structural MR imaging has limitations. vMRI does not directly evaluate Aβ and tau but, rather, provides an indirect assessment of neurodegeneration that occurs downstream from molecular pathology. Another limitation is that although certain patterns of volume loss are characteristic of different diseases (eg, entorhinal cortex atrophy in AD), the finding of medial temporal lobe atrophy by itself is nonspecific and can also be seen in other neurologic and psychiatric disorders. Therefore, vMRI of the medial temporal lobe structures, in isolation, cannot distinguish AD from hippocampal sclerosis or other neurodegenerative diseases such as frontotemporal dementia (FTD). Moreover, neuropathologic evidence demonstrates the presence of uncommon AD subtypes that spare the medial temporal lobes, especially in younger patients.29 Despite these weaknesses, given its capability for precise anatomic description with high reliability, analysis of MR imaging data across a wide range of scanner types/manufacturers, and the ability to efficiently generate normative databases from multicenter data, vMRI will undoubtedly play a significant role in decision making during the clinical evaluation of dementia. The optimal diagnostic and prognostic value of vMRI will be obtained when combined with clinical/cognitive testing and other markers including CSF and imaging measures of AD pathology.
Molecular Imaging and Fluid Biomarkers of Aβ Deposition
Within the last decade, a number of PET-based radiotracers have been developed to noninvasively assess for the presence of Aβ, of which the most extensively examined is 11C-labeled [N-methyl]-2-(4′-methylaminophenyl)-6-hydroxybenzothiazole (Pittsburgh Compound-B, PIB). Studies with transgenic mouse models and human brain sections indicate that PIB selectively binds to the fibrillar form of Aβ in neuritic plaques and cerebral amyloid angiopathy.30,31 In vivo, ante mortem PIB retention strongly correlates with in vitro, postmortem measures of fibrillar Aβ pathology in autopsy-confirmed AD but does not associate with NFTs, Lewy bodies, or other protein aggregates.32,33 In humans, the overall pattern of increased PIB retention mirrors the distribution of fibrillar Aβ plaques found at autopsy and involves the prefrontal, parietal, and lateral temporal cortices.34 A recent review suggests that the overwhelming majority of patients with AD and cognitively impaired patients who progress to AD are amyloid “positive.”35 Furthermore, approximately 24% of cognitively normal older adults older than 60 years also show increased cerebral PIB retention, and the prevalence of amyloid positivity is closely related to age and APOE ε4 carrier status.35 Together, these findings raise the possibility that amyloid imaging may yield positive results long before the appearance of cognitive symptoms, which, as discussed below, has both positive and negative consequences.
As either an alternative or adjunct to amyloid PET imaging, CSF sampling can also detect Aβ pathologic detail. Although most Aβ is produced in the brain and is secreted into the extracellular spaces of the brain, a fraction of central nervous system–produced Aβ diffuses into the CSF and is present in modest concentrations (approximately 10–15 ng/mL).36 CSF assessments measure the monomeric form of Aβ. Low CSF Aβ levels correlate strongly with increased PIB binding, intracranial plaque deposition, and total Aβ load, demonstrating the value of these CSF measurements as a marker of fibrillar Aβ pathologic findings.36 However, an important clinical consideration with CSF sampling is the need for lumbar puncture, an uncomfortable procedure that carries a small risk for morbidity.
Imaging Evaluation Strategy for MCI and AD
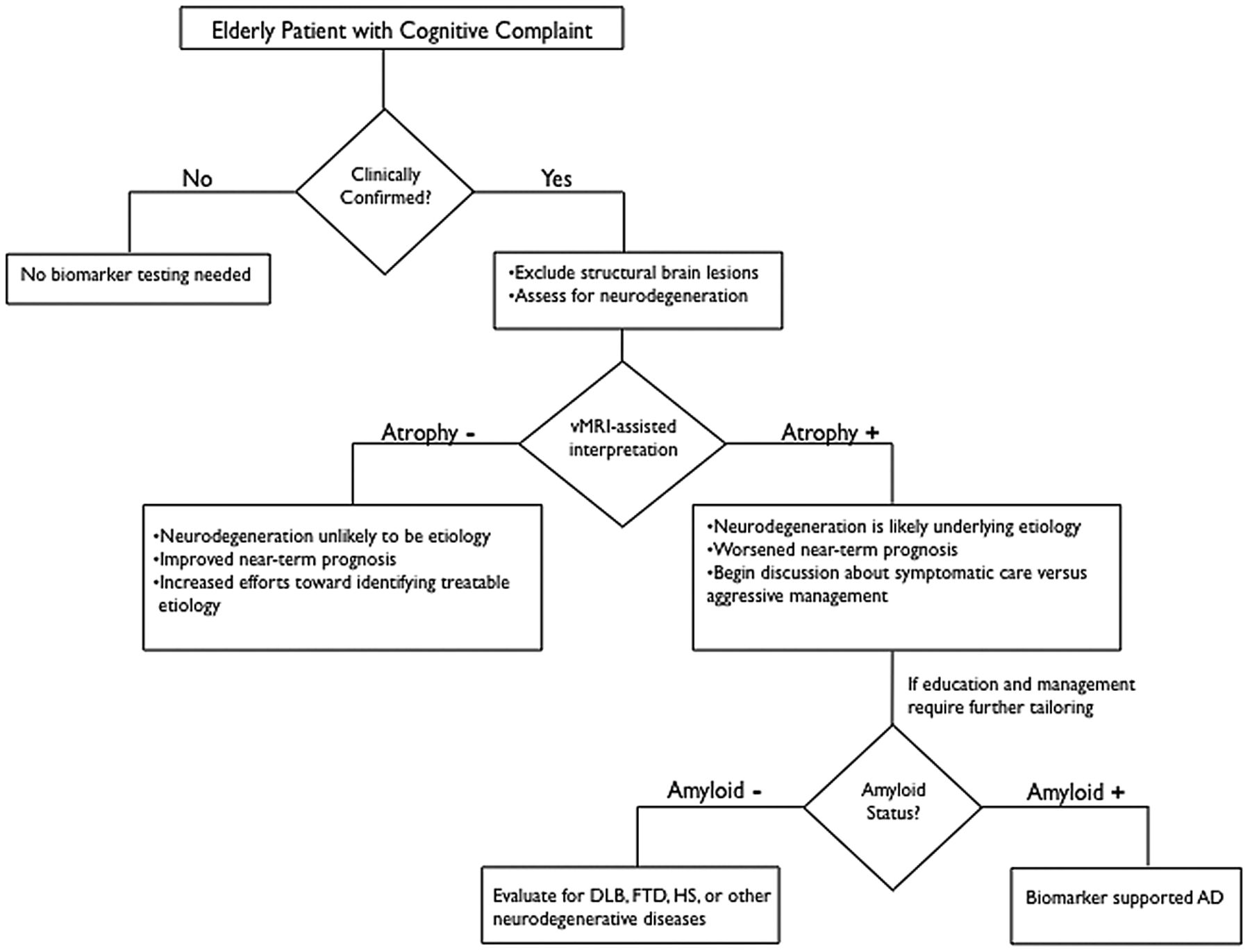
Clinical assessment of the elderly patient with a memory complaint usually begins with a mental status evaluation to objectively confirm the presence of the cognitive deficit. If the degree of cognitive decline is greater than expected for healthy aging and further information is needed to guide management, the determination of whether a neurodegenerative process underlies the cognitive complaint can help determine which further diagnostic method to use. Above and beyond exclusion of other conditions to explain cognitive deficits (eg, brain tumor, traumatic brain injury, infarctions, chronic hemorrhage, hydrocephalus, or encephalitis), structural MR imaging by use of vMRI techniques at this stage can be useful to document objective evidence of atrophy. As illustrated in Fig 2, vMRI can assist in supporting or contradicting a putative clinical diagnosis while providing an informative assessment of disease risk. The presence of reduced hippocampal volume provides support to the clinical impression that a neurodegenerative process contributes to the cognitive deficit but does not exclude the possibility of a congenitally small hippocampus or hippocampal damage from a prior insult. It is important to note that low hippocampal volumes do not specify whether the underlying cause is AD or other diseases such as FTD, dementia with Lewy bodies, or hippocampal sclerosis (Fig 2). Nevertheless, once a neurodegenerative cause is supported through clinical and radiologic evaluation, distinguishing among neurodegenerative disorders may benefit from supplemental testing for amyloid. This would likely be reserved for cases where additional tailoring of education or management is required and may be limited, as the more clinically relevant distinction is between benign or curable causes vs those with a near-term dire prognosis (Fig 2). It is important to note that in light of prior11 and recent37 clinical trial evidence that removing Aβ plaques by using immunotherapeutic methods may not halt the neurodegenerative process, amyloid testing to confirm AD as the underlying cause may prove most useful when therapies preventing downstream neurodegeneration become clinically available (Fig 2).

Recommended decision tree for evaluating the elderly patient with a cognitive complaint. DLB = Dementia with Lewy bodies; HS = hippocampal sclerosis. Figure adapted from McEvoy and Brewer.48
Challenges remain regarding the clinical application of vMRI in the patient with cognitive impairment. The difficulty in establishing normative ranges across a broad population of patients is a significant obstacle, but one that can be overcome by the availability of large data bases of images in cognitively normal elderly patients and patients with both MCI and AD enrolled in multisite, multinational initiatives such as the Alzheimer Disease Neuroimaging Initiative (ADNI) in North America and the AddNeuroMed Consortium in Europe (On-line Table). Because atrophy is not diagnostic of AD neuropathologically and because the hippocampus is affected by a broad array of disorders, the diagnosis of AD cannot rely on simple “cut points” or “thresholds” in hippocampal volume38,39 derived from studies of progression to AD dementia. Furthermore, the degree of abnormality, along with other radiologic features, including ex vacuo dilation of adjacent temporal horn and qualitative assessment of sulcal widening and cortical volume loss, will yield the impression of the presence or absence of neurodegeneration. It is important to note that the diagnosis of AD cannot be established by imaging alone; radiologic input serves to inform, rather than establish, an overall clinical impression.
Recommendations to use medial temporal atrophy on structural MR imaging among cognitively impaired patients have already been proposed by an international AD working group,40 and vMRI is one of the biomarkers recently incorporated into revised diagnostic criteria for AD, which noted that such biomarkers could serve “as optional clinical tools for use where available and when deemed appropriate by the clinician.”41 Consistent with these recently revised diagnostic guidelines for AD41 and MCI,42 by supporting the presence or absence of neurodegeneration, vMRI-based methods can also inform the likelihood of whether a patient with clinically confirmed memory loss will progress to dementia. The absence of vMRI-based brain atrophy diminishes the likelihood of neurodegeneration and increases the likelihood that a nonneurodegenerative, and potentially treatable, cause underlies the memory complaint. It is important to note that normal brain volumes for age, though not excluding the possibility of future neurodegeneration, can also be helpful to guide clinical management while providing a more accurate predictive prognosis. Normal hippocampal volumes confer a better near-term prognosis and can foster increased efforts toward finding a treatable cause for the memory impairment while providing needed, albeit cautious, reassurance to the patient and caregivers who will be anxious about being given a dire prognosis.
Amyloid Biomarkers in MCI and AD
The ability to specifically assess fibrillar Aβ pathology in vivo has generated considerable clinical excitement. Recently, the FDA has approved the fluorine-based amyloid tracer [F-18]florbetapir (Amyvid; Eli Lilly, Indianapolis, Indiana) for use in patients being evaluated for AD and other causes of cognitive decline (Fig 3). Furthermore, commercial CSF Aβ assays with established normative ranges for amyloid status are now clinically available (http://www.athenadiagnostics.com). However, as noted by the FDA, although a negative florbetapir (amyloid) scan result is inconsistent with a neuropathologic diagnosis of AD at the time of image acquisition, a positive florbetapir scan result does not establish a diagnosis of AD.43 Furthermore, elevated deposition of amyloid may occur in other neurologic conditions and is often present in healthy older adults with normal cognition. Recently, it has become increasingly evident that Aβ oligomers (eg, dimers, trimers, tetramers, and higher oligomers), rather than fibrillar Aβ plaques, represent the principal synaptotoxic form of amyloid that initiate the neurodegenerative process underlying AD. Insoluble Aβ fibrils, though serving as a reservoir for the neurotoxic oligomers, might themselves be relatively inactive.44 It is important to realize that neither CSF analytes nor amyloid imaging can detect the oligomeric form of Aβ.36 In a similar fashion, in cognitively normal older patients, although some studies have found a relationship between Aβ plaque deposition and neurodegeneration,45,46 recent studies have suggested that tau and other “downstream” markers of neuronal injury modulate the effect of Aβ on cognitive decline and brain atrophy.15–16,47 In addition, recent clinical trials with monoclonal antibodies (solanezumab and bapineuzumab) that target Aβ and promote its clearance from the brain demonstrate a minimal effect on disease trajectory modification in patients with mild or moderate AD: solanezumab showed marginal improvement in cognitive and functional decline, and bapineuzumab, though affecting fibrillar Aβ and tau levels, did not modify the disease trajectory.37 Taken collectively, this indicates that Aβ deposition precedes neurodegeneration and, in the absence of cognitive decline or brain atrophy, represents an elevated risk state in much the same fashion that hypercholesterolemia serves as a risk factor for heart disease in the absence of myocardial damage. Just as cholesterol levels would not be used to diagnose a myocardial infarction in the setting of chest pain, detecting amyloid deposition may be less valuable than markers of neuronal damage when determining the cause of ongoing memory impairment. Nevertheless, it is hoped that a future contribution of Aβ testing, from diagnostic and therapeutic perspectives, may be among cognitively normal adults before the onset of neurodegeneration.

Assessment of amyloid deposition by use of florbetapir (Amyvid). The axial PET image on the left shows normal preserved gray-white contrast with the cortical radioactivity less than the adjacent white matter (amyloid-“negative” scan). The axial PET image on the right demonstrates areas of decreased gray-white contrast with increased cortical radioactivity that is comparable to the radioactivity in the adjacent white matter (amyloid-“positive” scan). The florbetapir scan on the right was acquired on a patient with MCI who clinically progressed to dementia with a high biomarker probability of AD, as supported by this amyloid-positive scan and evidence of neuronal injury on structural MR imaging (Fig 1).
Role of Biomarkers in Guiding Clinical Management
Biomarker testing can help inform near-term prognosis by providing an objective assessment as to whether neurodegeneration is likely to be present. Whereas cognitive testing validates the patient or caregiver complaint that initiated the clinical visit, vMRI provides an orthogonal measure that is less overlapping with the patient complaint, thereby guarding against circularity in concluding that the cognitive problem is the result of AD. The presence of brain atrophy on vMRI, together with documented memory impairment confirmed by cognitive testing, suggests a prognosis of near-term decline and can prompt a discussion on evaluating the risk/benefit ratio for considering aggressive disease management vs symptomatic care (Fig 2). For patients and family members, these findings can help initiate a dialogue on future planning including determining the need for residential and driving support, involvement of a geriatric case manager, and financial decisions.
Evaluation with amyloid testing can prove useful once a neurodegenerative cause for cognitive decline has been established, especially in younger patients and in patients presenting with complaints atypical for AD. An amyloid test may be helpful for making a more informative dementia diagnosis (eg, AD vs FTD) in these patients, and can help guide the selection of medications for symptomatic management. As with vMRI, amyloid testing may also be of benefit to refine and tailor expectations while providing additional education to patients and caregivers.
The absence of brain atrophy on vMRI confers a better near-term prognosis and can provide cautious, but increased, optimism to physicians, patients, and caregivers. Although not excluding the possibility of future neurodegeneration, normal brain volumes can guide clinical management by prompting the physician to intensify efforts toward detecting a treatable cause for the patient's memory impairment (Fig 2). Such physician optimism is not lost on patients and may serve as needed reassurance to those patients with an inappropriately debilitating fear about progressing to dementia. Importantly, the intensified physician effort on behalf of patients whose complaints and cognitive impairments are incongruous with vMRI findings may lead to an improved likelihood of successful treatment and subsequent return of patients to normal cognitive function.
Potential Pitfalls with Biomarker Testing
In addition to valid concerns of added expense (Table 2), it is our opinion that biomarker assessment of patients without objective evidence of memory impairment could cause potential harm, as described by McEvoy and Brewer.48 For example, given the high frequency of nonspecific memory complaints in the general population and the high prevalence of amyloid positivity among the cognitively normal population, there is a significant chance that a patient's memory complaint is unrelated to intracranial Aβ deposition. A finding of elevated amyloid or low hippocampal volume might lead to inappropriate attribution of memory complaints to AD, circumventing a thorough work-up for other potentially treatable causes while exacerbating the debilitating worry that initially brought the patient to the clinic. Even in those patients where memory impairment is clinically confirmed, elevated amyloid levels do not assure that the cause of the impairment is AD. Amyloid positivity, in patients with objective memory decline, might lead to an overly simplistic attribution of memory complaints to AD and incomplete evaluations for modifiable causes of cognitive impairment. Finally, a negative amyloid test result is not necessarily a result to be celebrated because other neurodegenerative disorders should remain under consideration.
Disease progression markers in amnestic patients with MCI
Future Directions: Preclinical AD
Currently, there are no effective treatments that delay the onset or halt the progression from MCI to AD. There is increasing recognition that early intervention before the onset of neurodegeneration or clinical symptoms may represent the most effective treatment against AD,19 and a number of secondary prevention trials in preclinical older patients are currently underway. We believe that a screening strategy to assess dementia risk in cognitively normal adults could be useful if a meaningful therapy with minimal adverse effects becomes clinically available. Biomarker testing in asymptomatic patients is inherently controversial; therefore, we note that this evaluation strategy, though not currently applicable, may become relevant when or if meaningful preventive interventions are available.
Genetic, biochemical, and imaging evidence indicates that fibrillar Aβ pathologic change begins at least 15 years before the onset of clinical symptoms.49 Increasing levels of Aβ oligomers that progressively lead to plaque deposition are likely present at an even earlier age.50 These observations suggest that screening for the presence of amyloid should start in cognitively normal older adults (> 60 years old), similar to the current screening strategies for hypercholesterolemia or common cancers such as breast, colon, or prostate carcinoma. Although CSF concentrations of Aβ may become aberrant before amyloid imaging,49 additional factors such as the need to assess therapeutic response with time, clinical availability, and patient comfort should also be considered when determining whether to use fluid or imaging markers for amyloid status screening.
In cognitively normal older adults, a negative amyloid test result indicates a significantly lower risk for the development of AD. Because increased amyloid tracer uptake can also be seen with other conditions, such as cerebral amyloid angiopathy,32 a positive amyloid test result could be further evaluated with cognitive testing and, possibly, vMRI. Positive amyloid status along with the presence of progressive medial temporal lobe atrophy would suggest that the patient has entered the neurodegenerative phase of the disease process, which would change the risk/benefit calculation in considering more aggressive, less-benign medications that may become available. Although neuropathology remains the only way to definitively diagnose AD, available fluid and imaging markers supplement the physician toolbox for treating and educating patients and families worried about AD. As disease-modifying therapies are developed, this physician toolbox will likely evolve to further address the need for improved predictive prognosis and disease management in preclinical AD.
Acknowledgments
We thank Drs. Linda McEvoy, Dominic Holland, Gil Rabinovici, John Morris, Brad Hyman, Marilyn Albert, William Dillon, Bruce Fischl, Reisa Sperling, Keith Johnson, Clifford Jack, William Bradley, John Hesselink, Ole Andreassen, and Anders Dale for helpful input on an earlier draft of this manuscript. This study was supported by a grant from the National Institutes of Health (T32 EB005970).
Footnotes
All authors contributed equally to this study.
ICMJE Disclosures: Christopher Hess—RELATED: Grant: General Electric,* Comments: Research support; UNRELATED: Board Membership: General Electric,* Comments: Research support for projects related to dementia and ultrahigh-field MRI; Expert Testimony: Medicolegal consulting, Comments: occasional; Grants/Grants Pending: NIH.* James Brewer—UNRELATED: Consultancy: Lilly Biomarker Business Unit (Amyvid), Bristol-Myers Squibb, Avanir Pharmaceuticals, Comments: Advisory boards; Grants/Grants Pending: Janssen Alzheimer Immunotherapy,* General Electric Medical Foundation,* Comments: Support as Site PI of clinical trials- Janssen; Support for investigator initiated studies—General Electric; Payment for Lectures (including service on speaker bureaus): Lilly Biomarker Business Unit, Comments: Two lectures; Stock/Stock Options: Cortechs Laboratories, (NeuroQuant), Comments: Stock options less than 5%. *Money paid to institution.
Grant Support: Research support was provided by NIH grants NINDS K02 NS067427 (J.B.B.), NIA U01 AG10483 (J.B.B.), NIA P50 AG005131 (J.B.B.), NIA R01AG034062 (J.B.B.), NIH R01 HD072074–01 (C.P.H.), T32 EB005970 (R.S.D.), and General Electric Medical Foundation (J.B.B.).
Indicates open access to non-subscribers at www.ajnr.org
REFERENCES
- Received April 20, 2013.
- Accepted after revision April 22, 2013.
- © 2013 by American Journal of Neuroradiology




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Automated Segmentation of Hippocampal Volume: The Next Step in Neuroradiologic Diagnosis of Mesial Temporal Sclerosis
- Polygenic hazard score: an enrichment marker for Alzheimers associated amyloid and tau deposition
- Entorhinal Cortex: Antemortem Cortical Thickness and Postmortem Neurofibrillary Tangles and Amyloid Pathology